用于冠状动脉疾病的生物标记物的制作方法
相关申请的交叉参考无本发明涉及用于预测与微生物相关的疾病的风险的生物标记物和方法,特别是冠状动脉疾病(cad)或相关的心脏病。
背景技术:
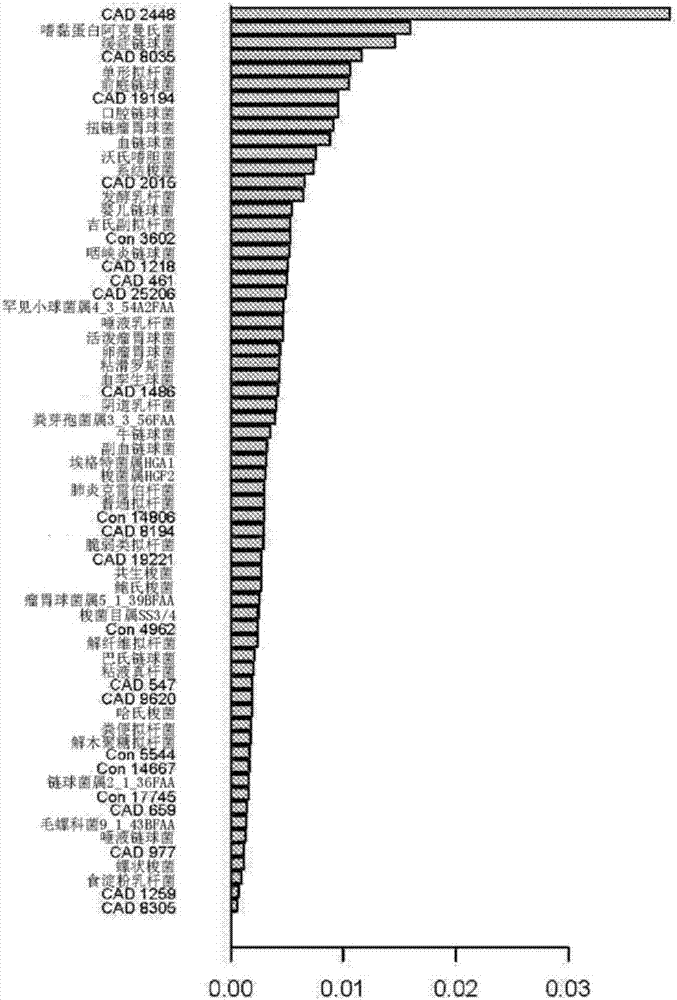
:冠状动脉疾病(cad)是指干扰向心(即心脏)肌肉或其任何部分输送充足血液的冠状动脉的任何异常状态。通常,cad由在动脉壁上斑块的积累(即动脉粥样硬化)引起,特别是在服务于心脏的大动脉和中等大小的动脉中。这些状态具有类似的病因、机制和治疗。cad代表全球死亡和发病的主要病因。cad的早期诊断不但会有助于预防死亡率,也降低手术干预的成本。用于检测cad的“金标准”是侵入性冠状动脉造影。然而,这是昂贵的,并且可能对患者造成风险。在血管造影之前,可以使用非侵入性诊断模式,例如心肌灌注成像(mpi)和ct-血管造影,然而这些具有包括辐射暴露、造影剂敏感性的并发症,并且仅适度地提高了阻塞性cad识别。目前的知识表明遗传、环境因素及其相互作用协同诱导复杂的表型和许多疾病。冠状动脉疾病(cad)作为最具影响力的复杂疾病之一,近年来已被gwas越来越多地研究,并且揭示了46种常见变异的10.6%的固有原因(ehret,g.b.等人,geneticvariantsinnovelpathwaysinfluencebloodpressureandcardiovasculardiseaserisk.nature478,103-109,通过引用并入本文中)。然而,我们对环境因素如肠道微生物的影响以及基因和微生物对疾病的贡献的知识仍需要进一步的研究。我们的“遗忘器官”肠道微生物群在许多方面对我们的健康起着至关重要的作用,如从食物摄取能量、产生重要的代谢物、促进免疫系统的发育和成熟、以及保护宿主免受病原体感染等。最近的研究表明,菌群生态失调、慢性炎症和代谢异常存在于某些代谢性疾病如糖尿病和肥胖的肠道中。大多数冠状动脉疾病的特征是炎症、氧化和脂质代谢,其可能与肠道微生物及其代谢物潜在相关。最近的研究表明,肠道微生物可以将红肉成分(l-肉碱、磷脂酰胆碱、胆固醇)代谢成tma,其将在肝脏中进一步氧化成tmao,从而在血管中引起氧化反应,引起炎症和脂质沉积,最终导致动脉粥样硬化和冠心病。同时,与健康受试者相比,症状性动脉粥样硬化患者肠微生物群表现出明显的异常(koeth,ra等人,intestinalmicrobiotametabolismofl-carnitine,anutrientinredmeat,promotesatherosclerosis.naturemedicine19,576-585,通过引用并入本文中)。这些研究表明肠道微生物的生态失调可能通过诱导人类代谢异常强烈影响冠状动脉疾病的发病机制。然而,动脉粥样硬化中肠道菌群失调的特征导致冠状动脉疾病患者的发病机制及其对代谢系统的影响仍然令人困惑。技术实现要素:本公开的实施例试图至少在某种程度上解决现有技术中存在的至少一个问题。本发明基于本发明人的以下发现:肠道微生物群的评估和表征已成为包括冠状动脉疾病(cad)的人类疾病的主要研究领域。为了对cad患者体内的肠道微生物内容物进行分析,本发明人进行了宏基因组关联研究(mgwas)的方案(qin,j.等人,ametagenome-wideassociationstudyofgutmicrobiotaintype2diabetes.nature490,55–60(20122),通过引用并入本文中),其基于来自165个个体的肠道微生物dna的深度鸟枪法测序。本发明人鉴别出并验证了65种cad相关的肠道微生物和4种优化的肠道微生物。为了利用肠微生物群的cad分类的潜在能力,本发明人基于65种cad相关的肠道微生物和4种优化的肠道微生物的随机森林模型计算疾病的概率。本发明人的数据提供了对与cad风险相关的肠道宏基因组的特征的洞察,用于未来研究肠道宏基因组在其它相关疾病中的病理生理学作用的范例,以及用于评估有风险患有此类病症的个体的基于肠道微生物群的方法的潜在有用性。在本公开的一个方面,提供了一种用于预测受试者体内与微生物群相关的疾病的生物标记物组,其由以下构成:肠道生物标记物,其包括嗜黏蛋白阿克曼氏菌(akkermansiamuciniphila)、脆弱类拟杆菌(bacteroidesfragilis)、鲍氏梭菌(clostridiumbolteae)、哈氏梭菌(clostridiumhathewayi)、系结梭菌(clostridiumnexile)、梭菌属hgf2(clostridiumsp.hgf2)、螺状梭菌(clostridiumspiroforme)、共生梭菌(clostridiumsymbiosum)、粪芽孢菌属3_3_56faa(coprobacillussp.3_3_56faa)、埃格特菌属hga1(eggerthellasp.hga1)、粘液真杆菌(eubacteriumlimosum)、血孪生球菌(gemellasanguinis)、肺炎克雷伯杆菌(klebsiellapneumoniae)、毛螺科菌9_1_43bfaa(lachnospiraceaebacterium9_1_43bfaa)、食淀粉乳杆菌(lactobacillusamylovorus)、发酵乳杆菌(lactobacillusfermentum)、唾液乳杆菌(lactobacillussalivarius)、阴道乳杆菌(lactobacillusvaginalis)、粘滑罗斯菌(rothiamucilaginosa)、活泼瘤胃球菌(ruminococcusgnavus)、卵瘤胃球菌(ruminococcusobeum)、瘤胃球菌属5_1_39bfaa(ruminococcussp.5_1_39bfaa)、扭链瘤胃球菌(ruminococcustorques)、咽峡炎链球菌(streptococcusanginosus)、牛链球菌(streptococcusinfantarius)、婴儿链球菌(streptococcusinfantis)、缓症链球菌(streptococcusmitis)、口腔链球菌(streptococcusoralis)、副血链球菌(streptococcusparasanguinis)、巴氏链球菌(streptococcuspasteurianus)、唾液链球菌(streptococcussalivarius)、血链球菌(streptococcussanguinis)、链球菌属2_1_36faa(streptococcussp.2_1_36faa)、前庭链球菌(streptococcusvestibularis)、罕见小球菌属4_3_54a2faa(subdoligranulumsp.4_3_54a2faa)、cvd1218、cvd1259、cvd1486、cvd19194、cvd19221、cvd2015、cvd2448、cvd25206、cvd461、cvd547、cvd659、cvd8035、cvd8194、cvd8305、cvd9620、cvd977、解纤维拟杆菌(bacteroidescellulosilyticus)、粪便拟杆菌(bacteroidesstercoris)、单形拟杆菌(bacteroidesuniformis)、普通拟杆菌(bacteroidesvulgatus)、解木聚糖拟杆菌(bacteroidesxylanisolvens)、沃氏嗜胆菌(bilophilawadsworthia)、梭菌目属ss3/4(clostridialessp.ss3/4)、吉氏副拟杆菌(parabacteroidesdistasonis)、con14667、con14806、con17745、con3602、con4962、con5544中的至少一种的肠道生物标记物,或具有包含seqidno:1至122009的至少部分序列的基因组dna的微生物,可选地,生物标记物组由表4中列出的物种中的至少一种构成,优选第表4中列出物种的至少10%、至少20%至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%、至少100%。优选口腔链球菌、血链球菌、缓症链球菌和婴儿链球菌中的至少一种。根据本公开的实施方案,肠道生物标记物包括如表5-1中所述的seqidno:1至122009中至少一个的至少部分序列。在本公开的另一方面,提供了用于预测受试者体内与微生物群相关的疾病的生物标记物组,其由以下构成:包括seqidno:1至122009中至少一个的至少部分序列的肠道生物标记物。根据本公开的实施方案,疾病为冠状动脉疾病或相关的心脏病。在本公开的另一方面,提供了用于确定权利要求1至4中任一项所述的基因标记物组的试剂盒,其包括用于pcr扩增并根据如下所述的dna序列设计的引物:包括seqidno:1至122009中至少一个的至少部分序列的肠道生物标记物。在本公开的另一方面,提供了用于确定上述基因标记物组的试剂盒,其包括一种以上根据如下所示的基因设计的探针:包括seqidno:1至122009中至少一个的至少部分序列的肠道生物标记物。在本公开的另一方面,提供了上述基因标记物组用于预测待测试的受试者体内冠状动脉疾病(cad)或相关疾病的风险的用途,包括:(1)从待测试的受试者收集样品;(2)确定步骤(1)中获得的样品中,上述的生物标记物组的每个生物标记物的相对丰度信息;(3)通过采用多元统计模型将待测试的受试者的每个生物标记物的相对丰度信息与训练数据集进行比较来获得cad的概率,其中cad的概率大于截止值表明待测试的受试者患有或有风险发展冠状动脉疾病(cad)或相关疾病。根据本公开的实施方案,训练数据集是采用多元统计模型基于多个患有cad的受试者和多个正常受试者的每个生物标记物的相对丰度信息构建的,可选地多元统计模型为随机森林模型。根据本公开的实施方案,训练数据集是矩阵,该矩阵中每行表示上述的生物标记物组的每个生物标记物,每列表示样品,每个单元表示样品中的生物标记物的相对丰度谱,样品疾病状态为矢量,其中1为cad,0为对照。根据本公开的实施方案,嗜黏蛋白阿克曼氏菌、脆弱类拟杆菌、鲍氏梭菌、哈氏梭菌、系结梭菌、梭菌属hgf2、螺状梭菌、共生梭菌、粪芽孢菌属3_3_56faa、埃格特菌属hga1、粘液真杆菌、血孪生球菌、肺炎克雷伯杆菌、毛螺科菌9_1_43bfaa、食淀粉乳杆菌、发酵乳杆菌、唾液乳杆菌、阴道乳杆菌、粘滑罗斯菌、活泼瘤胃球菌、卵瘤胃球菌、瘤胃球菌属5_1_39bfaa、扭链瘤胃球菌、咽峡炎链球菌、牛链球菌、婴儿链球菌、缓症链球菌、口腔链球菌、副血链球菌、巴氏链球菌、唾液链球菌、血链球菌、链球菌属2_1_36faa、前庭链球菌、罕见小球菌属4_3_54a2faa、cvd1218、cvd1259、cvd1486、cvd19194、cvd19221、cvd2015、cvd2448、cvd25206、cvd461、cvd547、cvd659、cvd8035、cvd8194、cvd8305、cvd9620、cvd977、解纤维拟杆菌、粪便拟杆菌、单形拟杆菌、普通拟杆菌、解木聚糖拟杆菌、沃氏嗜胆菌、梭菌目属ss3/4、吉氏副拟杆菌、con14667、con14806、con17745、con3602、con4962、con5544中每一种的相对丰度信息是基于seqidno:1至122009的相对丰度信息获得的。根据本公开的实施方案,训练数据集为表6-1、6-2、6-3、6-4、6-5中至少一者,并且cad的概率为至少0.5指示待测试的受试者患有或有风险发展冠状动脉疾病(cad)或相关疾病。在本公开的另一方面,提供了上述基因标记物组在制备用于预测待测试的受试者体内冠状动脉疾病(cad)或相关疾病的风险的试剂盒的用途,包括:(1)从待测试的受试者收集样品;(2)确定步骤(1)中获得的样品中上述的生物标记物组的每个生物标记物的相对丰度信息;(3)通过采用多元统计模型将待测试的受试者的每个生物标记物的相对丰度信息与训练数据集进行比较来获得cad的概率,其中cad的概率大于截止值表明待测试的受试者患有或有风险发展冠状动脉疾病(cad)或相关疾病。根据本公开的实施方案,训练数据集是采用多元统计模型基于多个患有cad的受试者和多个正常受试者的每个生物标记物的相对丰度信息构建的,可选地多元统计模型为随机森林模型。根据本公开的实施方案,训练数据集是矩阵,该矩阵中每行表示上述的生物标记物组的每个生物标记物,每列表示样品,每个单元表示样品中的生物标记物的相对丰度谱,样品疾病状态为矢量,其中1为cad,0为对照。根据本公开的实施方案,嗜黏蛋白阿克曼氏菌、脆弱类拟杆菌、鲍氏梭菌、哈氏梭菌、系结梭菌、梭菌属hgf2、螺状梭菌、共生梭菌、粪芽孢菌属3_3_56faa、埃格特菌属hga1、粘液真杆菌、血孪生球菌、肺炎克雷伯杆菌、毛螺科菌9_1_43bfaa、食淀粉乳杆菌、发酵乳杆菌、唾液乳杆菌、阴道乳杆菌、粘滑罗斯菌、活泼瘤胃球菌、卵瘤胃球菌、瘤胃球菌属5_1_39bfaa、扭链瘤胃球菌、咽峡炎链球菌、牛链球菌、婴儿链球菌、缓症链球菌、口腔链球菌、副血链球菌、巴氏链球菌、唾液链球菌、血链球菌、链球菌属2_1_36faa、前庭链球菌、罕见小球菌属4_3_54a2faa、cvd1218、cvd1259、cvd1486、cvd19194、cvd19221、cvd2015、cvd2448、cvd25206、cvd461、cvd547、cvd659、cvd8035、cvd8194、cvd8305、cvd9620、cvd977、解纤维拟杆菌、粪便拟杆菌、单形拟杆菌、普通拟杆菌、解木聚糖拟杆菌、沃氏嗜胆菌、梭菌目属ss3/4、吉氏副拟杆菌、con14667、con14806、con17745、con3602、con4962、con5544中每一种的相对丰度信息是基于seqidno:1至122009的相对丰度信息获得的。根据本公开的实施方案,训练数据集为表6-1、6-2、6-3、6-4、6-5中的至少一者,并且cad的概率为至少0.5指示待测试的受试者患有或有风险发展冠状动脉疾病(cad)或相关疾病。在本公开的另一方面,提供了诊断受试者是否具有与微生物群有关的异常状态或有风险发展与微生物群有关的异常状态的方法,包括:确定来自受试者的样品中上述的生物标记物的相对丰度,以及基于相对丰度确定受试者是否具有与微生物群有关的异常状态或者有风险发展与微生物群相关的异常状态。根据本公开的实施方案,所述方法包括:(1)从待测试的受试者收集样品;(2)确定步骤(1)中获得的样品中上述的生物标记物组的每个生物标记物的相对丰度信息;(3)通过采用多元统计模型将待测试的受试者的每个生物标记物的相对丰度信息与训练数据集进行比较来获得cad的概率,其中cad的概率大于截止值表明待测试的受试者具有或有风险发展冠状动脉疾病(cad)或相关疾病。根据本公开的实施方案,训练数据集是采用多元统计模型基于多个具有cad的受试者和多个正常受试者的每个生物标记物的相对丰度信息构建的,可选地多元统计模型为随机森林模型。根据本公开的实施方案,训练数据集是阵,该矩阵中每行表示根据上述的生物标记物组的每个生物标记物,每列表示样品,每个单元表示样品中的生物标记物的相对丰度谱,样品疾病状态为矢量,其中1为cad,0为对照。根据本公开的实施方案,嗜黏蛋白阿克曼氏菌、脆弱类拟杆菌、鲍氏梭菌、哈氏梭菌、系结梭菌、梭菌属hgf2、螺状梭菌、共生梭菌、粪芽孢菌属3_3_56faa、埃格特菌属hga1、粘液真杆菌、血孪生球菌、肺炎克雷伯杆菌、毛螺科菌9_1_43bfaa、食淀粉乳杆菌、发酵乳杆菌、唾液乳杆菌、阴道乳杆菌、粘滑罗斯菌、活泼瘤胃球菌、卵瘤胃球菌、瘤胃球菌属5_1_39bfaa、扭链瘤胃球菌、咽峡炎链球菌、牛链球菌、婴儿链球菌、缓症链球菌、口腔链球菌、副血链球菌、巴氏链球菌、唾液链球菌、血链球菌、链球菌属2_1_36faa、前庭链球菌、罕见小球菌属4_3_54a2faa、cvd1218、cvd1259、cvd1486、cvd19194、cvd19221、cvd2015、cvd2448、cvd25206、cvd461、cvd547、cvd659、cvd8035、cvd8194、cvd8305、cvd9620、cvd977、解纤维拟杆菌、粪便拟杆菌、单形拟杆菌、普通拟杆菌、解木聚糖拟杆菌、沃氏嗜胆菌、梭菌目属ss3/4、吉氏副拟杆菌、con14667、con14806、con17745、con3602、con4962、con5544中每一种的相对丰度信息是基于seqidno:1至122009的相对丰度信息获得的。根据本公开的实施方案,训练数据集为表6-1、6-2、6-3、6-4、6-5中至少一者,并且cad的概率为至少0.5指示待测试的受试者患有或有风险发展冠状动脉疾病(cad)或相关疾病。据认为,由于以下原因,65种cad相关的肠道微生物和4种优化的肠道微生物对于在早期阶段增加cad检测是有价值的。首先,本发明的标记物与常规标记物相比更特异和更敏感。第二,粪便分析保证准确性、安全性、可负担性和患者依从性。粪便样品是可运输的。因此,本发明涉及体外方法,其舒适且无创,因此人们将更容易参与给定的筛选程序。第三,本发明的标记物还可以用作对cad患者进行治疗监测的工具,以检测对治疗的响应。附图说明通过结合附图的以下描述,本公开的这些和其它方面和优点将变得清楚和更容易理解,其中:图1密度直方图示出了研究群组中鉴别出的所有基因的p值分布。水平线表示零假设下p值的分布。图2利用126种mlg标记物的随机森林模型中的65种最有判别性的mlg。棒长度表示变量(mlg物种)的重要性。图365种mlg随机森林模型的性能。165个样品(病例88,对照77)为训练集,并且其它86个样品(病例29,对照57)为测试集,用于验证假阴性率2/29和假阳性率12/57。图4肠道宏基因组中acvd相关标记物的鉴别。65种mlg随机森林模型的性能,将165个样品(88个病例和77对照)作为训练集(auc=98.17%)。两个外部曲线之间的面积表示95%ci形状。图5来自肠道宏基因组的acvd相关标记物的鉴别。4种mlg随机森林模型的性能,将165个样品(88个病例和77个对照)作为训练集(auc=85.86%)。两个外部曲线之间的面积表示95%ci形状。具体实施方式本文使用的术语具有与本发明相关领域的普通技术人员通常理解的含义。诸如“一”,“一个”和“所述”的术语不旨在仅指单数实体,而是包括可以用于说明的特定示例的一般类别。本文中的术语用于描述本发明的具体实施方案,但是它们的用法不限制本发明,除非在权利要求中概述。本发明在以下非限制性实施例中进一步举例说明。除非另有说明,份数和百分比以重量计,并且度数为摄氏度。对于本领域普通技术人员显而易见的是,这些实施例虽然指示本发明的优选实施方案,但仅以说明的方式给出,并且试剂可通过商业渠道获得。实施例1.鉴别用于评估冠状动脉疾病风险的生物标记物1.1样品收集来自包括88名动脉粥样硬化性心血管疾病(acvd)患者和77名对照受试者(训练集,表1)的165名南方中国受试者的粪便样品由广东省人民医院在2011年收集。acvd患者根据病理特征(冠状动脉造影)诊断并分类。要求受试者在医院收集新鲜粪便样品。将收集的样品置于无菌管中,立即储存在-80℃直至进一步分析。已经获得了完整的伦理批准,以及所有患者提供书面知情同意书。该研究由广东省人民医院伦理委员会批准。表1动脉粥样硬化性心血管疾病(acvd)病例和对照的基线特征。第四列报告了来自wilcoxon秩和检验的结果。参数病例(n=88)对照(n=77)p值年龄61590.294性别(男:女)68:1941:341.97e-05bmi24.5423.580.193注意:对于性别的信息,88个患者中的一个是未知的,并且77个对照中的两个是未知的。1.2dna提取将粪便样品在冰上解冻,并使用qiagenqiaampdnastoolmini试剂盒(qiagen公司)根据制造商的说明书进行dna提取。提取物用不含dna酶的rnase处理以消除rna污染。使用nanodrop分光光度计、qubit荧光计(具有quant-ittmdsdnabr测定试剂盒)和凝胶电泳测定dna的量。1.3粪便样品的dna文库构建和测序按照制造商的说明书(illumina公司)进行dna文库构建。本发明人采用与先前所述相同的工作流程来进行簇生成、模板杂交、等温扩增、线性化、封闭和变性,以及测序引物的杂交。本发明人为每个样品构建了具有插入片段大小为350bp的配对末端(pe)文库,随后进行高通量测序以获得长度为2x100bp的约3千万个pe读段。通过从illumina原始读段过滤掉具有不明确的“n”碱基、接头污染和人类dna污染的低质量读段,以及通过同时剪切掉读段的低质量末端碱基的低质量读段来获得高质量读段。发明人在illuminahiseq2000平台上从165个样品(88个病例和77个对照)中总共输出每个样品约4.77gb的粪便微生物群测序数据(高质量纯净数据)(表2)。表2宏基因组数据总结。第四列报告了来自wilcoxon秩和检验的结果。参数对照病例p值平均原始碱基(g)4.854.920.831去除低质量碱基后4.76(98.14%)4.79(97.36%)去除人类读段后4.73(97.53%)4.78(97.15%)0.8741.4宏基因组数据处理和分析1.4.1基因目录构建基因目录构建。采用与用于构建2型糖尿病基因目录相同的参数(qin,j.等人,ametagenome-wideassociationstudyofgutmicrobiotaintype2diabetes.nature490,55–60(2012),通过引用并入本文中),发明人分别采用soapdenovov1.06(li,r.等人,denovoassemblyofhumangenomeswithmassivelyparallelshortreadsequencing.genomeresearch20,265-272,doi:10.1101/gr.097261.109(2009),通过引用并入本文中)和genemarkv2.7(zhu,w.,lomsadze,a.&borodovsky,m.abinitiogeneidentificationmetagenomicsequences.nucleicacidsresearch38,e132,doi:10.1093/nar/gkq275(2010),通过引用并入本文中)对于165个样品的高质量读段进行了从头组装和基因预测。利用blat对所有预测的基因进行成对比对,其中超过其90%的长度可以以超过95%的同一性(不允许有间隙)比对到另一个的基因作为冗余被去除,产生出的非冗余基因目录包括4,537,046个基因(4.5m基因目录)。基因的分类分配。利用在公开的t2d论文(qin等人,2012,同上)中描述的内部流程(pipeline)进行预测基因的分类学分配。1.4.2数据配置文件构建基因谱。将这些4,537,046个基因及其在165个样品中的相对丰度的相关测量用于建立关联研究的基因谱(本发明人采用公开的t2d论文(qin等人,2012,同上)中描述的相同方法计算相对基因丰度。img物种和motu物种概况。以默认参数,将全部粪便纯净读段分别比对到来自imgv400(markowitz,v.m.等人,img:theintegratedmicrobialgenomesdatabaseandcomparativeanalysissystem.nucleicacidsresearch40,d115-d122(2012),通过引用并入本文中)的4,653个参考基因组和motu参考的79268个序列(unagawa,s.等人,metagenomicspeciesprofilingusinguniversalphylogeneticmarkergenes.naturemethods10,1196-1199(2013),通过引用并入本文中)。鉴别出1290种img物种(在至少10个受试者中共有的物种)和560种物种水平motu。1.4.3影响肠道微生物群基因特征的因素分析。本发明人采用置换多元方差分析(permanova)对4.5m参考基因目录的基因谱评估25种不同特征,包括cad状态、hdlc、chol、性别、fbg、高血压、apob、年龄、crea、ldlc、hba1c、apoa、tp、糖尿病、alb、trig、bmi、whr、lpa、hbdh、ckmb、ast、ck、probnp_e_、alt。本发明人使用在r软件中的“vegan”包中实施的方法进行分析,并且通过10,000次置换获得置换的p值。本发明人还用benjamini-hochberg方法在r软件中采用“p.adjust”来校正多重测试,以获得每个测试的q值。permanoa鉴别出与肠道微生物相关的两个重要因素(基于基因谱)(q<0.05,表3)。分析表明,cad和hdlc状态都是最强的相关标记物,支持疾病状态是影响肠道微生物群组成的主要决定因素。性别、年龄和一些cad临床指标如chol、fgb、高血压和apob也是重要因素。表3基于基因谱的欧氏距离分析(euclideandistanceanalysis)的permanova。所述分析用于测试临床参数和acvd状态是否对肠道微生物群有显著影响,其q值<0.05。1.4.4鉴别acvd相关标记物鉴别acvd相关基因。为了鉴别宏基因组模式和acvd之间的关联,在2.1m高频基因(在所有165个样品中存在于少于10个样品中的基因被移除)谱中使用双尾wilcoxon秩和检验。获得438,750个基因标记物(2.1m基因的20.48%),其在病例或对照中富集,其p值<0.01,fdr=2.23%(图1)。估计错误发现率(fdr)。代替顺序p值拒绝方法,本发明人应用在先前研究中提出的“q值”方法来估计fdr(storey,j.d.adirectapproachtofalsediscoveryrates.journaloftheroyalstatisticalsociety64,479-498(2002),通过引用并入本文中)。受试者工作特性(roc)分析。发明人应用roc分析来评估基于宏基因组标记物的acvd分类器的性能。然后,发明人使用r中的“proc”包来绘制roc曲线。1.4.5mlg的构建和acvd相关的mlg物种标记物的鉴别基于438,750个acvd相关标记物基因谱的126种mlg物种。本发明人使用438,750个基因标记物,采用在公开的t2d论文(qin等人,2012,同上)中描述的相同方法构建宏基因组连锁群(mlg)。所有438,750个基因通过将这些基因比对到imgv400中的4,653个参考基因组来注释。如果超过50%的组成基因注释到该基因组,则mlg被指定到该基因组,否则其被称为未分类。选择基因数>550的总共136个mlg基因组,将属于相同种类的这些mlg基因组分组以构建mlg物种,最终本发明人获得127种mlg物种。本发明人利用benjamini-hochberg调整对127种mlg物种进行了wilcoxon秩和检验,并且选择出126种mlg作为acvd相关mlg,其q<0.05。为了估计mlg物种的相对丰度,本发明人在去除5%最低和5%最高丰度基因后估计mlg物种的基因的平均丰度(qin等人,2012,同上)。总之,本发明人基于438,750个基因的分布和出现率(qin等人,2012,同上)构建了136个宏基因组连锁群(mlg>550个基因),94.8%的显著基因(p值<0.01)包括在mlg中。在ncbi数据库中注释136种mlg(每种>550个基因,>50%覆盖率并且q<0.05),并且将来自相同种类的mlg分组以获得126种mlg物种。65种mlg物种标记物鉴别。为了鉴别126种mlg物种标记物,发明人基于126种acvd相关mlg物种在2.10版r软件中使用“随机森林4.5-36”包。首先,本发明人按照“随机森林”方法给出的重要性将所有126种mlg物种分类(liaw,andy&wiener,matthew.classificationandregressionbyrandomforest,rnews(2002),vol.2/3p.18,通过引用并入本文中)。通过创建排列靠前的mlg物种的递增子集来构建mlg标记物组,从5种mlg物种开始,并在所有126种mlg物种处结束。对于每个mlg标记物组,发明人计算了我们的165个中国人群组中的假预测率。最后,选择出具有最低假预测率的65种mlg物种集作为mlg物种标记物(图2、表4和表5-1、5-2),其中假阴性(fn)率为6.81%(6/88)和假阳性(fp)率3.89%(3/77)(图3,训练集)。此外,发明人基于所选择的mlg物种标记物采用来自随机森林模型的疾病的oob(outofbag)预测概率绘制roc曲线(表6-1、6-2、6-3、6-4、6-5),并利用r软件包“proc”计算roc曲线下面积(auc)为98.17%(95%ci:96.6%-99.74%)(图4)。在65种mlg物种中,已知对照富集的mlg物种单形拟杆菌(q=4.21e-11)、普通拟杆菌(q=1.80e-09)和梭菌目属-ss3/4(q=1.68e-08)是已知的产scfa的细菌。大多数病例富集的mlg物种(共51种)是来自链球菌属(9/11mlg物种是口腔病原体)、梭菌目(6种mlg物种)、瘤胃球菌(4种mlg物种)和乳杆菌(4种mlg物种)的条件致病菌。粘滑罗斯菌天然地栖息在口腔和上呼吸道中,并且越来越多地被认为是与假体装置感染和心内膜炎相关的新兴的条件致病菌。鲍氏梭菌分离自人类粪便材料、血液和腹腔内脓肿,是革兰氏阳性病原体,并且可产生一些包括神经毒素的毒素,它在人类临床显著感染中遇到,其在自闭症儿童的平均数是对照儿童的46倍(p值=0.01)。血孪生球菌可以加强免疫缺陷患者的炎症。嗜粘蛋白阿克曼氏菌也在cad患者中富集。1.4.6鉴别acvd相关的img物种和motu物种。基于img物种谱和motu物种谱的img物种标记物和motu物种标记物的鉴别,本发明人鉴别出q<0.05(wilcoxon秩和检验与benjamini-hochberg调整)的acvd相关的img物种和motu物种。随后,如在mlg物种标记物选择中一样,采用随机森林方法选择img物种标记物和motu物种标记物。通过wilcoxon秩和检验和随机森林选择,具有roc98.52%的65种img物种和具有roc96.16%的15种motu物种也将cad患者与健康受试者明显分离(q<0.05;参见表7、8)。通过与65个mlg标记物重叠,发明人发现包括口腔链球菌、血链球菌、缓症链球菌和婴儿链球菌的口腔原病原体和嗜粘蛋白阿克曼氏菌在病例中显著分布。本发明人基于来自链球菌属(口腔链球菌、血链球菌、缓症链球菌和婴儿链球菌)的4种微生物作为生物标记物(表9)利用由随机森林模型的疾病的oob(outofbag)预测概率来绘制roc曲线并且使用r软件包“proc”计算roc曲线下的面积为85.86%(95%ci:80.24%-91.48%)(图5)。假阴性(fn)率为28.40%(25/88),并且假阳性(fp)率为20.77%(16/77)。实施例2.在另外86个个体中验证生物标记物为了验证生物标记物(即来自链球菌属的65个选定的mlg和4种微生物)的区分能力,本发明人使用另一个新的独立研究组,包括用作测试集的29个病例样品和57个对照样品(表10),并且也在广东省人民医院收集。表10.样品信息组病例对照总数测试集295786对于每个样品,提取dna并构建dna文库,然后如实施例1所述进行高通量测序。发明人通过使用来自该mlg的基因的相对丰度值,估计所有样品中mlg的相对丰度(qin等,2012,同上)。关于随机森林模型,在2.10版本r软件中使用“randomforest4.5-36”包,输入为训练数据集(即分别为表6-1、6-2、6-3、6-4、6-5或表9),样品疾病状态(训练数据集为矩阵,每行代表mlg;每列代表样品;每个细胞代表样品中mlg的相对丰度分布;实施例1中训练样本的样品疾病状态为矢量,1为cad,0为对照)和测试集(仅为测试集的mlg相对丰度分布)。然后,发明人使用r软件中的随机森林包的随机森林函数来构建分类,并且使用预测函数来预测测试集。输出为包含预测结果的矩阵(第一列“0”是健康概率;第二列“1”是cad的概率;截止值是0.5,并且如果cad的概率≥0.5,则受试者有风险患有cad的)。发明人使用65个选定的mlg重新进行随机森林,然后计算疾病的概率(表11,图3测试集)。在测试集(n=86,29个病例样品和57个对照样品)中测试模型并计算预测误差。假阴性率(fn)为6.89%(2/29)并且假阳性(fp)率为21.05%(12/57),并且roc曲线下面积为94.34%(95%ci:89.86%-98.83%)。此外,发明人使用来自4种链球菌属(口腔链球菌、血链球菌、缓症链球菌和婴儿链球菌)的微生物作为生物标记物,以测试分离cad患者和对照的能力(表11),发现阴性率(fn)率为17.24%(5/29)并且假阳性(fp)率为35.08%(20/57),并且roc曲线下面积为81.94%(95%ci:72.98%-90.9%)。表1165种mlg和4种mlg的预测结果因此,本发明人基于cad相关基因标记物的通过森林模型鉴别出并验证了65种cad相关的肠道微生物和4种优化的肠道微生物。并且发明人已经构建了基于这65种cad相关的肠道微生物和4种优化的肠道微生物来评估cad疾病的风险的方法。尽管已经示出和描述了示例性实施例,但是本领域技术人员应当理解,上述实施例不能被解释为限制本公开,并且可以在不脱离本公开的精神、原理和范围的情况下对实施例进行改变、替换和修改。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1