CXCR2拮抗剂用于预防和/或治疗化疗诱导的周围神经病变(CIPN)的用途的制作方法
本发明涉及CXCR2拮抗剂用于预防和/或治疗化疗诱导的周围神经病变(CIPN)的用途。
背景技术:
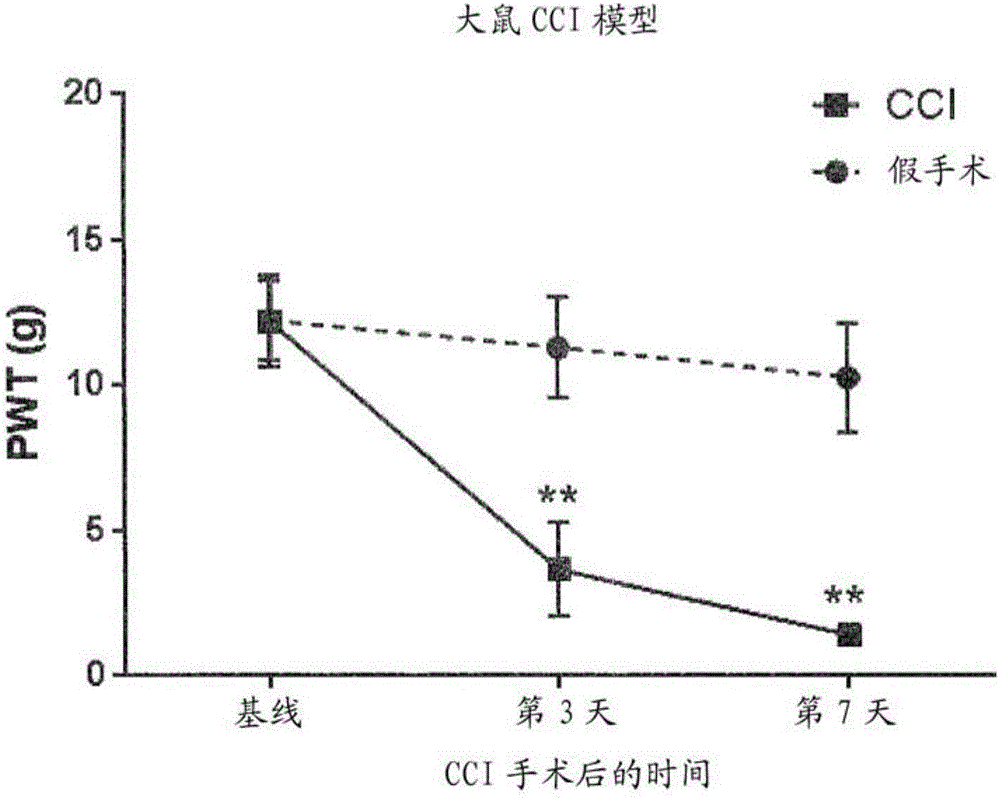
:CIPN被定义为接受化疗治疗方案的患者所经历的周围神经系统损伤。CIPN是许多化疗剂普遍主要的剂量限制性副作用,所述化疗剂包括铂化合物类(例如,奥沙利铂)、紫杉烷类、长春花生物碱类、沙利度胺和更新的药物,例如硼替佐米[Balayssac,ExpertOpin.DrugSaf.,10,407-417,2011)]。这代表了在许多不同的癌症中显著的治疗局限性,因为终末器官神经毒性和神经病可能对患者的生活质量具有极大影响且需要停止有效治疗。奥沙利铂已知能引起严重的急性和慢性周围神经病变[CassidyandMisset,Semin.Oncol.,29,11-20(2002);Extra,Semin.Oncol.,25,13-22(1998);Pasetto,Crit.Rev.Oncol.Hematol.,59,159-168(2006);和QuasthoffandHartung,J.Neurol.,249,9-17(2002)]。奥沙利铂在重复治疗后在晚期引起大鼠坐骨神经中有髓纤维的退化[Kawashiri,Eur.J.Pain,15,344(2011)],且在小鼠模型中用奥沙利铂慢性治疗诱导背根神经节(DRG)神经元收缩和有髓纤维的轴突损伤[Renn,Mol.Pain,7,29(2011);Cavaletti,Eur.J.Cancer,37,2457(2001)]。啮齿类中感觉终点的早期改变已在奥沙利铂模型中发现,导致对热(Neuropharmacology,79(2014)432-443;Pain,101(2003)65–77)和机械刺激(Neuropharmacology,79(2014)432-443;BrainResearch,784(1998)154–162)的敏感性增加,其分别使用冷的丙酮刺激和vonFrey细丝。例如,包含奥沙利铂的治疗方案(例如每2周85mg/m2)在95%的患者中产生即时性‘冷’敏感的暂时性感觉异常和肢体肌肉痉挛,其发展成无运动神经元参与的对称性轴突感觉远端原发性神经病[Argyriou,CancerTreatmentReviews,43,368-377(2008)]。在背根神经节(DRG)及其感觉神经元中的奥沙利铂摄取和铂积累是奥沙利铂神经毒性的主要决定因素(Jong,J.Pharmacol.Exp.Ther.,338(2):537-47(2011)。此外,炎性级联激活在CIPN的引发和进展中起作用,其中免疫细胞渗入受损的神经元环境中[Wang,Cytokine,59,3-9(2012)]。促炎性趋化因子受体CXCR2表达于感觉神经元且其配体已参与调节钠和钾电流的增加,其控制神经元兴奋性[Wang,Mol.Pain,24,38(2008);Yang,Mol.Pain,5,26(2009)]。在外周神经元损伤中,啮齿类中CXCR2+促炎性分泌型免疫细胞的聚集也已知涉及一些急性和持续性疼痛状态,其被CXCR2拮抗作用阻断[Manjavachi,Eur.J.Pain,14,23-31(2010);Kiguchi,J.Pharmacol.Exp.Ther.,340,577-587(2012);Stadtmann&Zarbock,Front.Immunol.,3,263(2012)]。CXCR2配体已经显示出调节参与伤害感受过程的TRPv1通道的功能[Dong,Neurosci.Bull.,28,155-164(2012)]并刺激钙内流和感觉神经元中疼痛介导肽降钙素基因相关肽(CGRP)的释放(Qin,J.Neurosci.Res.,82,51-62(2005))。人外周神经外植体和施万细胞培养物表达[Ozaki,NeuroReport,19,31-35(2008)]并分泌CXCR2促炎性细胞因子,例如IL-8[Rutkowski,J.Neuroimmunol.,101,47-60(1999)],其在糖尿病和酒精性神经病以及在长度依赖性小纤维神经病中显著升高[AboElAsar,Cytokine,59,86-93(2012)];(Michalowska-Wender,FoliaNeuropathol.,45,78-81(2007);Neurology,74,1806(2010)]。所述神经元CXCR2受体系统也已显示调节髓鞘再生(re-myelination)[Veenstra&Ransohoff,J.Neuroimmunol.,246,1-9(2012)]和突触可塑性(Xiong,J.Neurosci.Res.,71,600-607(2003)过程,其控制神经元的通信。所述CXCR2受体及其配体在结肠直肠癌中也被上调并已参与药物抗性[Acharyya,Cell,150,165-178(2012)]、肿瘤生长、血管形成、癌细胞增殖和嗜中性粒细胞聚集到肿瘤微环境[Verbeke,Cytokine&GrowthFactorReview,22,345-358(2012)]。目前尚无用于预防和/或治疗CIPN的获批治疗选择。技术实现要素:根据第一方面,本发明提供了用于预防和/或治疗CIPN的CXCR2拮抗剂。根据第二方面,本发明提供了在需要的人中预防和/或治疗CIPN的方法,其包括给药CXCR2拮抗剂。根据第三方面,本发明提供了CXCR2拮抗剂在制备用于预防和/或治疗CIPN的药物中的用途。附图简述图1是显示在大鼠中慢性压迫性损伤(chronicconstrictioninjury,CCI)后对缩足阈值(PWT)的影响的图。图2是显示小鼠中慢性压迫性损伤后对PWT的影响的图。图3是显示在大鼠中慢性压迫性损伤后坐骨神经中对mRNA水平的影响的一系列图。图4是显示在小鼠中慢性压迫性损伤后坐骨神经中对mRNA水平的影响的一系列图。图5是显示在大鼠中单次注射奥沙利铂后对PWT的影响的图。图6是显示在小鼠中单次注射奥沙利铂后对PWT的影响的图。图7是显示在大鼠中单次注射奥沙利铂后左侧坐骨神经中对mRNA水平的影响的一系列图。图8是显示在小鼠中单次注射奥沙利铂后左侧坐骨神经中对mRNA水平的影响的一系列图。图9是显示化疗诱导的神经病变的大鼠模型中式(A)化合物的预防性治疗后对PWT的影响的图。图10是显示用式(A)化合物或其载体治疗后大鼠坐骨神经中对奥沙利铂减少的神经传导速度的影响的图。图11是显示用式(A)化合物或其载体治疗后对大鼠体重的影响的图。图12是一组三个显微照片,显示在奥沙利铂治疗的大鼠中用式(A)化合物治疗后对坐骨神经的髓鞘的厚度的影响。图13是显示在奥沙利铂治疗的大鼠中用式(A)化合物治疗后对坐骨神经的髓鞘的厚度的影响的柱状图。发明详述迄今为止还没有公开CXCR2调节能对周围神经病变(特别是CIPN)提供有效的预防和/或治疗方案。本文所述的机理临床研究方案的证明,预期能提供有关人的证据,即CXCR2的有效和选择性的拮抗剂可为CIPN的有效治疗方案。该机理临床研究的证明是在患有结肠直肠癌的患者中进行,该患者即将经历包含奥沙利铂的化疗治疗方案的周期。这种使用CXCR2拮抗剂的治疗将在化疗周期中和化疗周期前后间歇地给予。该方案保证最高的CXCR2抑制同时具有最高的全身奥沙利铂暴露量。该研究将在每个化疗周期之前、之中和之后测量多种相关的终点。主要终点是使用神经生理学跟踪技术测量周围神经兴奋性。由含有奥沙利铂的治疗方案引起的感觉神经兴奋性(SE)的急性增加可比建立的神经生理学神经传导速度标记物更早地检测到,所述治疗方案被认为是由节点轴索电压门控钠通道(nodalaxonalvoltage-gatedsodiumchannel)的功能障碍引起的[Krishnan,MuscleNerve,32,51-60(2005)],并且该急性增加已经被建议用来预测奥沙利铂诱导的周围神经病变的发生和严重性[Park,Brain,132,10,2712-23(2009)]。在奥沙利铂的第5个周期之前SE从基线增加15%,使得在奥沙利铂治疗的第12个周期结束时检测到80%的病例具有中度至重度神经毒性。而且,机理临床研究方案的证明已被设计以提供以下证据,即有效且选择性的CXCR2拮抗剂能有效预防和/或治疗CIPN,其通过捕获在奥沙利铂周期过程中与感觉神经兴奋性的变化相关的患者结果信息。此外,CXCR2拮抗剂的临床前研究已证明,抑制CXCR2对奥沙利铂的抗增殖作用没有不利影响[Ning,Mol.CancerTher.,11,1353-1364(2012)]。机理研究的证明也将监测包含奥沙利铂的治疗方案对癌症相关的终点的有效性。因此根据第一方面,本发明提供了用于预防和/或治疗CIPN的CXCR2拮抗剂。如本文所述,术语CXCR2拮抗剂是指抑制CXCR2受体的激动剂-介导的响应的化合物。本文使用的术语预防是指完全阻止神经损伤和/或相应的神经功能障碍(通过神经生理跟踪技术测量)或减缓神经损伤和/或神经功能变化的进展。应理解预防包括a)化疗剂对健康神经没有造成损伤或基本上没有损伤的情况;b)受损的神经(例如由早期化疗周期引起)不被化疗剂进一步损伤或基本上不被损伤的情况;和c)与任何先前的化疗周期相比,对神经的损伤(由早期化疗循环的累积效应引起)减少的情况。本文使用的术语治疗是指逆转或部分逆转CIPN,其中神经损伤和/或相应的神经功能障碍(通过神经生理跟踪技术测量)被修复或逆转,导致神经功能的改善,从而减少神经病的症状。CIPN是独特的神经病。在大多数其它神经病(如糖尿病性神经病和酒精性神经病)中,在损害/神经损伤已发生一段时间后出现神经性症状。经历化疗的患者中神经病发生的可能性,特别是经历奥沙利铂化疗的神经病的高发生率[DecisionResorcesLLC,KantarHealth,OncologyAnalyser,IntrinsiqDatabase;DeGramont,J.Clin.Oncol.,18,2938-2947(2000)],意味着在给予化疗剂之前或同时给予CXCR2拮抗剂具有防止神经病急性发生和防止慢性持久的神经性症状的建立的潜能。机理临床研究的证明将通过调节神经兴奋性措施及其建议的患者结果的预测性翻译的证据而测量该预防性方面。在一个实施方案中,本发明提供用于预防CIPN的CXCR2拮抗剂。如上所述,已知许多化疗剂引起CIPN,例如铂化合物(例如,奥沙利铂)、紫杉烷、长春花生物碱、沙利度胺和硼替佐米。在一个实施方案中,该CIPN由包括含铂的化疗剂的化疗引起。在一个实施方案中,该CIPN由包括奥沙利铂的化疗引起。在一个实施方案中,该CIPN由含铂的化疗剂引起。在另一实施方案中,该CIPN由奥沙利铂引起。CXCR2拮抗剂用于本发明的适合性可通过根据标准药物实践评价其功效、选择性、吸收、代谢、药代动力学、毒性等而测定。CXCR2拮抗剂的功效可使用容易获得的测试方法测定,如以下实验部分描述的测试a)、b)、c)和d)。测试a)测量人内源性激动剂IL-8与重组表达的人CXCR2受体的放射性标记的结合。测试b)测量重组表达的人CXCR2受体的功能活性,其通过报告基因构建体在受体自身下游测量。测试c)测量重组表达的人CXCR2受体的功能活性,其通过报告基因构建体在受体自身下游测量。测试d)测量全血中天然表达的人CXCR2的细胞功能活性,其通过流式细胞仪细胞分选方案在受体自身下游测量。在每个测试中,对激动剂介导的终点的抑制决定拮抗剂的效力。对于给定的拮抗剂,效力可以根据所使用的测定法而不同。然而,本领域药理学人员能够容易地比较不同测试的结果来确定效力。在一个实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于7.0,其在CXCR2受体结合测试中测量。在另一实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于7.8,其在受体结合测试中测量。在一个实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于7.0,其在测试a)中测量。在另一实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于7.8,其在测试a)中测量。在一个实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pA2值大于或等于8.0,其在测试b)中测量。在另一实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pA2值大于或等于8.2,其在测试b)中测量。在一个实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于8.0,其在测试c)中测量。在另一实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于8.8,其在测试c)中测量。在一个实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于5.0,其在测试d)中测量。在另一实施方案中,该CXCR2拮抗剂对抗CXCR2受体的pIC50值大于或等于5.4,其在测试d)中测量。CXCR2拮抗剂对CXCR2受体相对于CXCR1受体的选择性可使用测试a)和e)测定。选择性比例可容易通过对具体涉及的受体的相应IC50值的比例由本领域技术人员测定。在一个实施方案中,该CXCR2拮抗剂对CXCR2的选择性是CXCR1的大于或等于29倍。在另一实施方案中,该CXCR2拮抗剂对CXCR2的选择性是CXCR1的大于或等于50倍。在一个实施方案中,该CXCR2拮抗剂对CXCR2的选择性是CXCR1的大于或等于100倍。口服生物利用度是指口服给药的药物达到体循环的比例。确定药物口服生物利用度的因素为溶解度、膜渗透性、代谢稳定性和主动转运体的可能参与。通常,筛选级联法首先使用体外研究鉴定可能的倾向,然后推进至体内评估以确定口服生物利用度。溶出度,即胃-肠道(GIT)中的水性内含物对药物的溶解作用,可从体外溶解度实验预测,其在合适的pH进行以模拟GIT。在一个实施方案中,该CXCR2拮抗剂的最小溶解度为10μg/ml。溶解度可通过本领域的标准步骤测定,如Adv.DrugDeliv.Rev.,23,3-25(1997)中所述。膜渗透性是指化合物穿过生物膜的比率。亲脂性是预测的关键性质且通过使用有机溶剂和缓冲液的体外LogD7.4测量值定义。在一个实施方案中,CXCR2拮抗剂的LogD7.4为-2至+4,更优选-1至+3。LogD7.4可通过本领域已知的标准步骤测定,如J.Pharm.Pharmacol.,42:144(1990)中所述。代谢稳定性涉及在吸收和消除过程中GIT或肝代谢化合物的能力。测试系统如微粒体、肝细胞等可预测代谢倾向性。在一个实施方案中,该CXCR2拮抗剂在测试系统中显示代谢稳定性,其与低至中度的肝萃取相称。测试系统和数据操作的实例描述于Curr.Opin.DrugDisc.Devel.,4,36-44(2001);DrugMet.Disp.,28,1518-1523(2000)。由于上述过程的相互作用,可通过在临床前物种中的体内实验进一步评估药物是否潜在可被人口服生物利用。在这些研究中通过口服和静脉内途径给予化合物确定绝对生物利用度。在动物中评估口服生物利用度的实例可参见DrugMet.Disp.,29,82-87(2001);J.MedChem.,40,827-829(1997);DrugMet.Disp.,27,221-226(1999)。合适用于本发明的CXCR2拮抗剂公开于欧洲专利公开EP1558259B1和EP2009992B1。这些公开的内容,特别是其中的权利要求的治疗活性化合物的通式和实施例化合物,以其整体在此引入作为参考。在一个实施方案中,用于本发明的CXCR2拮抗剂选自:式(A)的1-(4-氯-2-羟基-3-(哌嗪-1-基磺酰基)苯基)-3-(2-氯-3-氟苯基)脲式(B)的1-(4-氯-2-羟基-3-(哌啶-3-基磺酰基)苯基)-3-(3-氟-2-甲基苯基)脲式(C)的1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲式(D)的1-(4-氯-3-((1,4-二甲基哌啶-4-基)磺酰基)-2-羟基苯基)-3-(2-氯-3-氟苯基)脲式(E)的(R)-1-(4-氯-2-羟基-3-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯基)-3-(2-氯环戊-2-烯-1-基)脲式(F)的(R)-1-(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲式(G)的1-(4-氯-2-羟基-3-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲式(H)的1-(4-氯-3-((反-3-(二甲基氨基)环丁基)磺酰基)-2-羟基苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲式(I)的(R)-1-(4-氯-3-((1,1-二氟乙基)磺酰基)-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲式(J)的1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲式(K)的1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲和式(L)的1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲或其药学上可接受的盐。式(A)的化合物可根据欧洲专利EP1558259B1实施例1所述的方法制备。式(B)的化合物可根据欧洲专利EP2009992B1实施例1所述的方法制备。式(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)和(L)的化合物可根据以下所述的方法制备。还可参见国际专利申请PCT/EP2015/061618的制备。在另一实施方案中,所述CXCR2拮抗剂为式(A)的1-(4-氯-2-羟基-3-(哌嗪-1-基磺酰基)苯基)-3-(2-氯-3-氟苯基)脲或其药学上可接受的盐在另一实施方案中,所述CXCR2拮抗剂为式(B)的1-(4-氯-2-羟基-3-(哌啶-3-基磺酰基)苯基)-3-(3-氟-2-甲基苯基)脲或其药学上可接受的盐在另一实施方案中,所述CXCR2拮抗剂为式(C)的1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲或其药学上可接受的盐在另一实施方案中,所述CXCR2拮抗剂为式(J)的1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲或其药学上可接受的盐该CXCR2拮抗剂可单独给药,但通常以与合适的药物赋形剂、稀释剂或载体的混合物给药,该药物赋形剂、稀释剂或载体根据预期给药途径和标准药物实践而选择。在通过合适的途径给药于患者前,该CXCR2拮抗剂通常,但不必须,配制为药物组合物。因此,在另一方面,本发明提供了一种药物组合物,其包含a)CXCR2拮抗剂,和b)用于预防和/或治疗CIPN的一种或多种药学上可接受的赋形剂。本发明的药物组合物可以以散装形式进行制备和包装,其中将本发明化合物分配然后给药至所述患者(例如粉末或糖浆)。或者,可将本发明药物组合物制备和包装成药物剂型,其中每种物理上分散的药物剂型含有本发明化合物。因此,在另一方面,本发明提供了包含本发明药物组合物的药物剂型。对于平均体重范围为约65至80kg的人受试者,每个分散的药物剂型通常含有5mg至100mg的本发明化合物。本领域技术人员能够容易确定体重在该范围之外的受试者所需的剂量水平,如儿童和老年人。本发明组合物将通常被配制成适于通过所需给药途径给药至患者的药物剂型。例如,药物剂型包括适于以下的那些:(1)口服给药,例如片剂、胶囊、小胶囊、丸剂、锭剂、粉末、糖浆、酏剂、混悬液、溶液、乳液、囊剂和扁囊剂;(2)胃肠外给药,例如无菌溶液、混悬液、植入物和用于重构的粉末;(3)透皮给药,例如透皮贴剂;(4)直肠和阴道给药,例如栓剂、阴道栓剂和泡沫;(5)吸入和鼻内给药,例如干粉、气雾剂、混悬液和溶液(喷雾和滴剂);(6)局部给药,例如乳膏剂、软膏剂、洗剂、溶液、糊剂、滴剂、喷雾剂、泡沫和凝胶;(7)眼部给药,例如滴剂、软膏剂、喷雾剂、混悬液和嵌入物;(8)口腔和舌下给药,例如锭剂、贴剂、喷雾剂、滴剂、咀嚼片和片剂。在一个实施方案中,该剂型适用于口服给药。在一个实施方案中,该组合物适用于口服给药。如本文所述,术语"药学上可接受的赋形剂"是指不与CXCR2拮抗剂明显反应,且不导致对CXCR2拮抗剂的治疗活性具有不希望的作用的物质。药学上可接受的赋形剂将根据所选具体药物剂型而变化。此外,可根据具体功能选择合适的药学上可接受的赋形剂,使它们适用于该组合物。例如,可根据其促进均一药物剂型的产生的能力选择一些药学上可接受的赋形剂。可根据其促进稳定的药物剂型的产生的能力选择一些药学上可接受的赋形剂。可根据一旦将化合物或本发明化合物给药至患者便促进从一个器官或机体部分携带或运输所述化合物或本发明化合物至另一器官或机体的另一部分的能力选择一些药学上可接受的赋形剂。可根据其提高患者顺应性的能力来选择一些药学上可接受的赋形剂。可根据其促进CXCR2拮抗剂以适当的速率释放以治疗该病症的能力来选择一些药学上可接受的赋形剂。用于适合口服给药的组合物的药学上可接受的赋形剂包括:稀释剂和填料(如乳糖、蔗糖、葡萄糖、甘露醇、山梨醇、玉米淀粉、马铃薯淀粉、预胶化淀粉、纤维素、微晶纤维素、硫酸钙、和磷酸氢钙);粘合剂(如淀粉、玉米淀粉、马铃薯淀粉、预胶化淀粉、明胶、阿拉伯胶、藻酸钠、海藻酸、黄蓍胶、瓜尔胶、聚乙烯吡咯酮、纤维素和羟丙基甲基纤维素);崩解剂(如淀粉、聚乙烯聚吡咯烷酮、羟乙酸淀粉钠、交联羧甲醚纤维素、海藻酸和羧甲基纤维素钠);润滑剂(如硬脂酸、硬脂酸镁、硬脂酸钙、和十二烷基硫酸钠);助流剂(如滑石和胶体二氧化硅);成粒剂;包衣剂;湿润剂;溶剂;共溶剂;悬浮剂;乳化剂;增甜剂;调味剂;掩味剂;着色剂;抗结剂;润湿剂;螯合剂;增塑剂;增粘剂;速率调节剂;抗氧化剂;防腐剂;稳定剂;表面活性剂;和缓冲剂。本领域技术人员将理解,一些药学上可接受的赋形剂可起到一种以上的功能,并且根据该赋形剂在所述制剂中存在多少以及该制剂中存在什么其他成分可起到替代的功能。在一个实施方案中,适合口服给药的剂型如片剂、胶囊、囊片和丸剂,可配制以用于立即、延迟、修饰、持续、脉冲或控制释放本发明的化合物。本领域技术人员具有本领域的知识和技能,使它们能够确定合适的药学上可接受的赋形剂以适当的量用于CXCR2拮抗剂中。此外,本领域技术人员可以获得许多资源,其描述了药学上可接受的赋形剂并可用于选择合适的药学上可接受的赋形剂。实例包括Remington'sPharmaceuticalSciences(MackPublishingCompany)、TheHandbookofPharmaceuticalAdditives(GowerPublishingLimited)和TheHandbookofPharmaceuticalExcipients(theAmericanPharmaceuticalAssociationandthePharmaceuticalPress)。本发明药物组合物可使用本领域技术人员已知的技术和方法进行制备。本领域常用的一些方法描述于Remington'sPharmaceuticalSciences(MackPublishingCompany)。以下配制例仅为示例性的且不意欲限制本发明的范围。根据本发明,该CXCR2拮抗剂预防和/或治疗通过给药化疗剂引起的CIPN。因此,该CXCR2拮抗剂可与化疗剂组合给药。在本发明一个实施方案中,该用于预防和/或治疗CIPN的CXCR2拮抗剂与一种或多种主要化疗剂组合,该活性剂选自以下列表:铂化合物(例如,奥沙利铂)、紫杉烷、长春花生物碱、沙利度胺和硼替佐米。在另一实施方案中,该主要化疗剂为奥沙利铂。通常,化疗剂与额外的药剂(例如,其他化疗剂、血管生成抑制剂,止吐剂)组合给药,其为实现患者顺应性抗癌治疗提供了增加的治疗选择,包括治疗难治性群体。在本发明另一实施方案中,该CXCR2拮抗剂与以下一起给药:a)主要化疗剂和b)选自以下的额外的药剂:贝伐单抗、伊立替康、卡培他滨、西妥昔单抗、帕尼单抗、瑞戈非尼和Ziv-阿柏西普。如果给予活性剂的组合,则它们可同时、分开或相继给药。在一个实施方案中,该CXCR2拮抗剂在给药化疗剂之前或同时给药。应理解,本发明提供了以下其它方面的基础。上述第一方面中的具体实施方案延伸至这些方面:在需要的人中预防和/或治疗CIPN的方法,包括给药CXCR2拮抗剂;CXCR2拮抗剂在制备用于预防和/或治疗CIPN的药物中的用途;用于预防和/或治疗CIPN的药物组合,其包含CXCR2拮抗剂和另一种上文定义的活性剂;在需要的人中预防和/或治疗CIPN的方法,包括给药包含CXCR2拮抗剂和另一种上文定义的活性剂的药物组合;药物组合在制备用于预防和/或治疗患者CIPN的药物中的用途,该药物组合包含CXCR2拮抗剂和另一种上文定义的活性剂;用于预防和/或治疗CIPN的试剂盒,该试剂盒包含:a)包含CXCR2拮抗剂的第一药物组合物;b)包含另一种上文定义的活性剂的第二组合物;和c)用于该第一和第二组合物的容器;和试剂盒在制备用于预防和/或治疗CIPN的药物中的用途,该试剂盒包含:a)包含CXCR2拮抗剂的第一药物组合物;b)包含另一种上文定义的活性剂的第二组合物;和c)用于该第一和第二组合物的容器。实验部分a)CXCR2和CXCR1的受体结合测试[125I]IL-8(人重组体,IM249)获自AmershamCorp.,ArlingtonHeights,IL,比活性为2000Ci/mmol。所有其它化学品为分析级。高水平的重组人CXCR1和CXCR2受体分别在中国仓鼠卵巢(CHO)细胞中表达,如Holmes,等人,Science,1991,253,1278所述,将其在此引入以进行本测试。中国仓鼠卵巢膜根据Haour,等人,J.Biol.Chem.,249pp2195-2205(1974)制备,将其在此引入以进行本测试,除了将均化缓冲液改变为包含1mMMgS04、0.5mMEDTA(乙二胺四乙酸)、1mMPMSF(α-甲苯-磺酰氟)、2.5mg/L亮抑酶肽和0.1mg/ml抑肽酶的40mMTris-HCLpH7.5。膜蛋白质浓度使用Bio-Rad试剂使用牛血清白蛋白作为标准而测定。所有结合测试使用闪烁亲近测定法(SPA测试)进行,其使用小麦胚芽凝集素珠在96-孔微孔板(optiplate96,Packard)格式中。该CHO-CXCR2和CHO-CXCR1膜在结合缓冲液中用珠子预培养;20mMBisTris丙烷,pH8,其包含25mMNaCl、1mMMgSO4、0.1mMEDTA,在测试前在4℃保持30分钟。化合物以终浓度的20倍在100%DMSO中稀释(最终1nM至1000nM和5%DMSO)。该测在包含结合缓冲液、用小麦胚芽凝集素珠预处理的膜、多种浓度的化合物、5%DMSO、0.04%CHAP、0.0025%BSA和0.225nM[125I]IL-8的0.1ml反应缓冲液中进行。该96-孔板在振动平台上培养1小时。在培养结束时将板以2000RPM旋转5分钟,且在TopCount计数器中计数。认为显示pIC50值>5的化合物在测试中具有活性。式(A)的化合物对CXCR2受体的pIC50值为7.8且对CXCR1受体的pIC50值为5.5。如该测试所测量,因此式(A)的化合物对CXCR2的选择性是CXCR1的188倍。式(B)的化合物对CXCR2受体的pIC50值为7.9且对CXCR1受体的pIC50值为6.0。如该测试所测量,因此式(B)的化合物对CXCR2的选择性是CXCR1的78倍。b)钙动员测试稳定表达CXCR2和Gα16的CHO-K1细胞在DMEM/F12(HAM's)1:1,w/10%FCS(热灭活),w/2mML-谷氨酰胺,w/0.4mg/mlG418中生长至80%融合,同时在5%CO2培养箱中维持在37℃。在进行测定之前24小时,收获细胞并在96孔、黑色壁、透明底板(PackardView)中铺板(每孔40,000个细胞),并返回到CO2培养箱中。在测定当天,将化合物在100%DMSO中连续稀释至所需测试浓度的300倍。从细胞中吸出生长培养基,并用100μL负载培养基(具有Earl's盐w/L谷氨酰胺,0.1%BSA(来自SeriologicalsCorp.的BovunlinarCohenFractionV)的EMEM,4uMFluo-4-乙酰氧基甲基酯荧光指示剂染料(Fluo-4AM,来自MolecularProbes)和2.5mM丙磺舒)在37℃在CO2培养箱中培养1小时。抽吸负载培养基并用100μL具有Earl's盐w/L-谷氨酰胺、0.1%明胶和2.5mM丙磺舒的EMEM更换,并且再培养10分钟。将300x的在DMSO中的连续稀释的化合物(3μL)转移到含有297微升KRH(120mMNaCl,4.6mMKCl,1.03mMKH2PO4,25mMNaHCO3,1.0mMCaCl2,1.1mMMgCl2,11mM葡萄糖,20mMHEPES(pH7.4))w/2.5mM丙磺舒和0.1%明胶的96孔板(现在化合物为3x浓度)。从细胞中吸出培养基,并用KRHw/2.5mM丙磺舒、w/0.1%明胶洗涤细胞3次。将具有0.1%明胶的KRH(100μL)w/2.5mM丙磺舒加入孔中,然后将50μL在KRHw/2.5mM丙磺舒和0.1%明胶中的3x化合物加入孔中并在37℃在CO2培养箱中培养10分钟。将板置于FLIPR(FluorometricImagingPlateReader,MolecularDevices,SunnyvaleCalif)上用于如前所述的分析[SarauetaI.,Mol.Pharmacol.,Sep.,56(3),657-63(1999)]。针对化合物的各浓度测定由1.0nMIL-8(CXCR2的EC80浓度)诱导的最大人IL-8诱导的Ca2+迁移的百分比,并且IC50计算为抑制由1.0nMIL-8诱导的最大响应的50%的测试化合物的浓度。如该测试所测量,式(A)的化合物对CXCR2受体的pA2值为8.4。如该测试所测量,式(B)的化合物对CXCR2的pA2值为8.2。c)CXCR2Tango测试该测试测定在含有与TEV蛋白酶位点和Gal4-VP16转录因子相连的重组人CXCR2的稳定细胞系(Invitrogen)中,受体CXCR2的配体诱导的活化。配体与所述受体的结合导致抑制蛋白(用蛋白酶标记)募集至所述受体并触发转录因子的释放受限。所述转录因子进入细胞核并活化报告基因的转录。化合物抑制CXCR2活化的能力通过测量所述报告基因的活性而间接评估。将一小瓶冷藏保存的细胞从液氮中取出并在37℃的水浴中轻轻搅拌以迅速解冻。收集该细胞内含物并以约200,000个细胞/ml的密度再混悬于测试培养基中。将所有的测试化合物以10mM的浓度溶于DMSO中,然后连续稀释以在384-孔测试板(Greiner781090)中产生10-点剂量响应曲线,其使用Echo(Labcyte)浓度-响应程序(50nl/孔)。向所述无细胞、未刺激和阳性对照中装入50nl/孔纯的DMSO以确保所有测试板的DMSO浓度恒定。使用具有标准盒的Multi-dropCombi(Thermo),在含化合物的测试板中将50μl测试培养基加至无细胞对照的各孔中;将50μl无hCXCL1(CXCR2配体)的细胞混悬液加至未刺激的对照孔(每孔约10,000个细胞);并将50μl具有80nMhCXCL1的细胞混悬液加至该测试板的剩余孔中(每孔约10,000个细胞)。将该细胞在37℃/5%CO2培养过夜。使用具有小管盒的Multi-dropCombi(Thermo)将10μl的6x底物混合物(LiveBLAzerTM-FRETB/G底物(CCF4-AM),目录号K1096,购自Invitrogen,Inc.)加至各孔并将该板在室温避光培养2h。然后将该板在EnVision上使用一个激发通道(409nm)和两个发射通道(460nm和530nm)进行读取。通过将除去背景的蓝色发射值除以除去背景的绿色发射值来计算各孔的蓝/绿发射比率(460nm/530nm)。所述剂量响应曲线基于S型剂量-响应模型。所有的比率数据基于各板的阳性对照(hCXCL1)的最大发射比率和阴性对照(DMSO)的最小发射比率进行标准化。如该测试所测量,式(A)的化合物对CXCR2受体的pIC50为9.2。如该测试所测量,式(B)的化合物对CXCR2受体的pIC50为8.8。如该测试所测量,式(C)的化合物对CXCR2受体的pIC50为9.0。如该测试所测量,式(D)化合物对CXCR2受体的pIC50为8.9。如该测试所测量,式(E)化合物对CXCR2受体的pIC50为8.8。如该测试所测量,式(F)的化合物对CXCR2受体的pIC50为9.7。如该测试所测量,式(G)的化合物对CXCR2受体的pIC50为9.0。如该测试所测量,式(H)的化合物对CXCR2受体的pIC50为9.0。如该测试所测量,式(I)的化合物对CXCR2受体的pIC50为9.4。如该测试所测量,式(J)的化合物对CXCR2受体的pIC50为9.0。d)人全血测试版本i)通过静脉穿刺在含有225微升0.25MEDTA的10ml注射器中由训练有素的医疗人员从健康同意的成年人获得人血液并填充7ml血液(在整个CXCL1刺激期间保持在37℃)。将测试化合物溶于100%DMSO中至10mM的浓度,并在1.2%DMSO中稀释至测定浓度的12倍(DMSO终浓度为0.1%)。测试:将100μl全血加入含有10微升1.2%DMSO(测定浓度为0.1%)或10微升溶解在1.2%DMSO中的化合物的测试管或96孔U底透明板中(测试浓度范围0.1μM至10μM),然后在37℃培养10分钟,随后加入10μl的DPBS中的0.1%BSA(阴性对照)或10μl的120nMCXCL1(GROα,PreproTech,Inc)(溶于0.1%BSA的DPBS溶液中)(测试浓度为10nM,其为CXCL1的EC80),并在轻轻搅拌下在37℃培养另外10分钟,然后置于冰上,通过加入250μlCellFix或250μl3.7%v/v多聚甲醛(BDBiosciences)终止测试。一分钟后,将50μl固定血加入含有10μl抗CD11b-FITC(BeckmanCoulter)和5μl抗人CD16-PE(DakoCytomation)的管中,轻轻混合,且将总样品加入500μl冷DPBS中。在使用LSR流式细胞仪(Becton-Dickinson)或FACSCantoII分析之前,将细胞在4℃用10μlLDS-751(MolecularProbes)核酸染色剂染色10分钟,允许细胞仪阈值设置排除红细胞。使用CellQuest软件或FACsdiva进行分析。通过顺序门控测定嗜中性粒细胞群;首先在侧向散射对前向散射图中对所有粒细胞进行门控,然后对粒细胞群(CD16+嗜中性粒细胞,CD16-嗜酸性粒细胞)中的CD16+(PE荧光)细胞进行门控。评估每个样品的嗜中性粒细胞群体的平均FITC荧光,因为其直接涉及CD11b含量,即CXCR2激活的生物标志物。测定每个样品的平均荧光。阳性对照定义为用10nMCXCL1(无抑制剂)刺激的样品的荧光值。阴性对照定义为载体处理的样品(无CXCL1,无抑制剂)的荧光值。从每个样品中减去平均阴性对照值。然后将所有样品相对于平均阳性对照进行归一化。如该版本的测试中所测量的,式(A)的化合物对CXCR2受体的pIC50为5.7。如该版本的测试中所测量的,式(B)的化合物对CXCR2受体的pIC50为6.4。如该版本的测试中所测量的,式(D)化合物对CXCR2受体的pIC50为5.4。版本ii)通过静脉穿刺从知情的成人中抽取血液并倒入含有抗凝血剂肝素(10uL/mL血液)的Sterilin管中。将所有测试化合物溶于DMSO至4mM并在板中以1:3连续稀释,得到10点剂量响应曲线。然后将该化合物在PBS[磷酸盐缓冲液(无氯化钙和氯化镁)]中稀释100倍,之后使用FX将1uL分配于96U型底的Costar板上。这么做是为了将最终的DMSO浓度降低至0.25%并在加入血液后以10uM的最终测试浓度筛选化合物。使用多通道移液管将10μL血液转移至化合物板,轻轻敲打并在37℃培养15分钟。15分钟后,将刺激物GROα在0.1%BSA(牛血清白蛋白)-PBS中稀释成100nM并将5μL分配至整个板上,使最终浓度为33nM。轻轻敲打平板并在37℃再培养15分钟。将该板置于冰上1分钟,然后加入10μL由BioLegend(地址:CambridgeBioscience,MunroHouse,TrafalgarWay,BarHill,Cambridge,UK)的CD11b-FITC(40ug/mL)和购自BDPharmingen(地址:TheDanbyBuilding,EdmundHalleyRoad,OxfordSciencePark,Oxford,UK)的CD16-PE组成的抗体混合物(储备浓度100倍测试浓度,将2mL储备体积以1:5稀释)。将该板在冰上避光1小时。然后将该细胞使用200uL/孔的1xFACS(流式激活细胞分选(FlowActivatedCellSorting))裂解溶液(Becton和Dickinson-BD)进行固定并在冰上避光20分钟。将该板以1600rpm离心5分钟并用200uL冰冷的PBS再混悬。将这个步骤重复两次并在最终步骤中将该板用50uL冰冷的-PBS再混悬,用于流式细胞检测分析。将样品在Becton和Dickinson(BD)-AcurriC6流式细胞仪上,使用HyperCyt采样装置(IntelliCyt)以2uL/秒的流速运行。监测在嗜中性粒细胞中CD11b的上调并使用侧向散射和CD16表达的组合进行鉴定。如该版本的测试中所测量的,式(J)的化合物对CXCR2受体的pIC50为7.4。如该版本的测试中所测量的,式(D)化合物对CXCR2受体的pIC50为5.8。该值类似于该测试版本ii)中测量的式D的化合物获得的pIC505.4,因此测试d)的版本i)和ii)的结果是相当的。e)GPCR抑制蛋白测试本发明的某些化合物还由DiscoveRXCorporation(42501AlbraeStreet,Fremont,CA94538,UnitedStates)在他们的GPCR抑制蛋白(Arrestin)测定中测试,以确定它们对包括CXCR2和CXCR1的一组受体的活性。根据标准方法从冷冻库中扩增PathHunterCHO细胞系。将细胞以20μL的总体积接种到白壁384孔微量培养板中,并在测试前在37℃培养适当时间。将细胞与拮抗剂预培养,随后在EC80浓度下进行激动剂刺激。进行样品储液的中间稀释以在测试缓冲液中产生5X样品。将5μL5x样品加入细胞中,并在37℃或室温培养30分钟。载体浓度为1%。将5μL的测试缓冲液中的6XEC80激动剂加入到细胞中,并在37℃或室温培养90或180分钟。通过单次添加12.5或15μL(50%v/v)PathHunter检测试剂混合物,随后在室温培养1小时产生测试信号。在用用于化学发光信号检测的PerkinElmerEnvisionTM仪器产生信号后读取微孔板。使用CBIS数据分析套件(ChemInnovation,CA)分析化合物活性。使用下式计算抑制百分比:%抑制=100%×(1-(测试样品的平均RLU-载体对照的平均RLU)/(EC80对照的平均RLU-载体对照的平均RLU))。如该测试所测量,式(J)的化合物对CXCR2受体的pIC50值为8.3且对CXCR1受体的pIC50值为5.4。如该测试所测量,式(J)的化合物对CXCR2的选择性是CXCR1的730倍。如该测试所测量,式(D)化合物对CXCR2受体的pIC50值为7.0且对CXCR1受体的pIC50值为5.6。如该测试所测量,式(D)化合物对CXCR2的选择性是CXCR1的29倍。f)奥沙利铂和式(A)的化合物在结肠直肠癌细胞系增殖测试中的作用细胞增殖测试:细胞以不同细胞密度接种于96-孔板(Costar):HCT116,500个细胞/孔;Caco-2,1000个细胞/孔。培养基的总体积为90μl/孔。细胞在37℃和5%CO2培养器中培养24小时。奥沙利铂和式(A)的化合物浓度系列进一步用DPBS稀释。奥沙利铂在HCT116细胞系实验中的终浓度来自500μM的3倍稀释,15种浓度和一个对照。The终浓度sof奥沙利铂在Caco-2细胞系实验中的终浓度来自55.5μM的3倍稀释,9种浓度和一个对照。式(A)的化合物的终浓度为8.5μM。5μL奥沙利铂连同5μl载体或式(A)的化合物一起添加至培养基且再在37℃和5%CO2培养器中培养72小时。细胞增殖速率通过添加100μlCellTiter-GloTM(Promega)根据制造商的说明进行测量。培养10分钟后,将细胞裂解物转移至OptiPlate-384孔,通过BioTekSynergyTM4Hybrid微板读取器使用Gen5软件记录发光。所有数据点一式五份生成。进行三个独立的实验。数据分析:pIC50值通过软件GraphPadPrism5使用S形浓度-响应(可变斜率)计算。所有数据表示为来自三个独立实验确定的平均值的平均值±标准误差。通过从三个独立实验确定的单因素ANOVA方差分析(GraphPadPrism5)分析单独的式(A)化合物(8.5μM)对Caco-2和HCT116细胞系的基底增殖的影响。在这些分析中包括从奥沙利铂浓度响应曲线和载体对照的整体产生的所有数据,以允许相关统计学评价奥沙利铂的确定的生物效应的背景下单独的式(A)化合物的作用。结果:奥沙利铂显示在Caco-2和HCT116细胞系中对细胞增殖的浓度-依赖性抑制,其中pIC50值分别为5.923±0.047和6.049±0.016。在浓度为8.5μM的式(A)的化合物的存在下,这些奥沙利铂抑制细胞增殖的效力不受影响,其中pIC50值分别为5.95±0.07和6.05±0.02。在Caco-2和HCT116细胞系中;单独的式(A)化合物存在基底增殖的非显著性降低,分别为2.7±2.1%和8.9±0.7%。此外,这些影响小于在各个奥沙利铂浓度响应曲线中观察到的生物相关统计学差异。因此,式(A)的化合物(8.5μM)单独对两种细胞系的基底细胞增殖没有影响。g)慢性压迫性损伤啮齿类中的临床前基因表达神经性疼痛的慢性压迫性损伤(CCI)模型涉及具有四个结扎的坐骨神经的单侧松散连接。这导致出现痛觉过敏,异常性疼痛和自发性疼痛(异位放电),部分由外周中炎性细胞的募集和脊髓中小胶质细胞和星形胶质细胞的活化引起。认为该模型模拟在临床中观察到的神经性疼痛的一些症状和病因(Bennett和Xie,Pain198833(1):87-107(1988);Field等,Pain83(2):303-11(1999)。在CCI模型中已经观察到啮齿动物中感觉终点的早期改变,导致对热(Neuropharmacology,79(2014)432-443;Pain,101(2003)65-77)和机械刺激(Neuropharmacology,79(2014)432-443;BrainResearch,784(1998)154-162)的敏感性增加,分别使用冷丙酮刺激和vonFrey细丝。雄性SpragueDawley大鼠(Harlan,UK.200-250克)在四个一组的标准笼养和实验室条件下饲养,自由获取食物(5CR4,Purina)和水(放置在测试箱中时除外),以12/12光/暗循环。从到达日提供环境丰富化,并在星期一(城堡),星期三(房子)和星期五(管)改变以防止自残。将雄性C57BL6小鼠(Harlan,UK,6-7周龄)放在标准的笼中,实验室条件和5个一组的环境富集中,自由获得食物(5CR4,Purina)和水(放置在测试箱中时除外),以12/12光/暗循环。行为部分:基线测试:所有动物在手术前进行机械性异常性疼痛的行为测试,以确定基线缩足阈值。对于大鼠研究,将对用von-Frey纤毛刺激的最后两个(共三个)的基线缩足阈值(PWT)的平均值作为基线。对于小鼠研究,在两个分开的天(第-1天和第0天)收集基线数据。手术过程:在与氧气混合的异氟烷麻醉(3:1,1L/min)下,将左后腿在大腿中部水平剃毛,并使用手术刀穿过皮肤进行切口。通过使用一对锋利的剪刀进行初始切开来解剖股二头肌层,然后使用一对钝剪刀将其扩大。暴露常见的坐骨神经,并且在坐骨神经周围绑扎3根(用于小鼠)或4根(用于大鼠)疏松绑扎的聚丙烯缝线(对于小鼠)或铬肠线(SMI)(对于大鼠),每个之间具有1mm的间距。然后将神经返回到肌肉层下面,使用可吸收缝线(Vicryl)闭合伤口。假手术动物进行相同的程序,只是神经未结扎。在手术后3天开始行为测试,在第3天和第7天采取机械响应阈值。在CCI手术治疗后的第1天、第3天和第7天收集来自DRG和坐骨神经的组织。在第1天和第3天使用单独的手术制备的动物组进行组织收集,并在第3天进行行为评估。通过放置在适当大小的标记管中并放置在液氮中将所有组织速冻。静态机械(触觉)异常性疼痛:大鼠:将动物置于具有0.5cm2网格的升高的网底平台上,并将倒置的有机玻璃容器置于各动物之上。在初始15-20分钟适应/习惯期后进行测试。使用校准的(力;g)von-Frey单丝(触摸测试感觉评估器;ScientificMarketingAssociates)实现缩足阈值的测量,其应用后爪的足底表面约8秒,用足够的力以引起细丝的稍微弯曲。缩足阈值通过增加和减少刺激强度来确定,并使用Dixon的上下方法估计[Dixon,Annu.Rev.Pharmacol.Toxicol.,20,441-62(1980);Chaplan等人,J.Neurosci.Methods,Jul,53(1),55-63(1994)]。仅认为对刺激的立即快速缩回反应(或退缩)代表阳性反应。小鼠:通过使用应用于后爪的足底表面的校准(力;g)von-Frey单丝(Touch-TestSensoryEvaluator;ScientificMarketingAssociates),通过测量缩足阈值来评估静态机械性异常性疼痛。在试验前将动物置于凸起的金属网上的单独的Perspex盒中30-40分钟。以1秒开1秒关的方案依次施加一系列渐变的vonFrey纤毛(0.07,0.16,0.4,0.6和1g),重复10次。将每根纤毛垂直地施加到爪的腹侧表面的中心,直到它略微弯曲。施加到动物后爪以在10个轨迹中诱导5个响应的力将被记录为PWT。仅认为对刺激的立即快速缩回反应(或退缩)代表阳性反应。在连续两天(第-1天和第0天)评估和在手术后第3天和第7天重新评估PWT,以监测机械性异常性疼痛的发展。统计分析:行为数据通过GraphpadPrism使用常规双因素ANOVA方差分析进行分析,“治疗”作为受试者之间的效应,且“天”作为受试者内的效应。使用Bonferroni方法进行事后分析。p值<0.05被认为是统计学显著的。mRNA表达:QuantStudioOpenArray用于评估CCI对触觉行为终点后NPY(神经肽Y)、CXCR2及其同族配体CXCL1的表达水平的影响。解剖出大鼠和小鼠的坐骨神经并在液氮中快速冷冻。将组织在-80℃冷冻储存或在干冰上储存,直到处理用于CXCR2和CXCL1的RNA分析。ActB、GAPDH、HPRT1和MAPK6用作内部测试对照和标准品,并且NPY用作CCI的对照。使用Q-PCR测量以下基因。测试用于制备预扩增池。大鼠的具体测试基因符号Taqman测试IDActBRn00667869_m1CXCL1Rn00578225_m1CXCR2Rn00567841_m1GAPDHRn01775763_g1HPRT1Rn01527840_m1MAPK6Rn00581152_m1NPYRn01410145_m1小鼠的具体测试基因符号Taqman测试IDActBMm01205647_g1CXCL1Mm04207460_m1CXCR2Mm00438258_m1GAPDHMm99999915_g1HPRT1Mm01545399_m1MAPK6Mm00727050_s1NPYMm03048253_m1总RNA分离:对于每个组织样品,通过根据制造商的说明在RocheMagnaLyser上的GreenBead管(Roche)中的MagNAPureLC裂解缓冲液(Roche)中将组织匀浆来制备一个RNA样品,并使用RocheMagnaPureRNA分离系统(Roche)根据制造商手册中的说明进行总RNA分离。将样品以50μL等分试样洗脱,并在80℃储存直至需要。RNA定量和完整性评估:根据制造商的手册,在LifeTechnologiesQubit上定量每个总RNA样品。使用AgilentPico试剂盒(AgilentTechnologies)根据制造商的手册在Agilent2100生物分析仪上评估总RNA样品的代表性样品的完整性。如果RIN(RNA完整性数)为6或更大,则认为RNA是可接受的质量。第一链cDNA合成:使用SuperscriptVilo试剂盒(LifeTechnologies)从50ng的每种总RNA合成第一链cDNA。每个RNA用无RNase的水补足至11μL,并向每个RNA样品中加入14μL主混合物。主混合物基于下表制成:组分体积5xViloRT缓冲液5μL10xVilo酶混合物2.5μL无核酸酶的水6.5μL最终体积RT主混合物14μL将样品在DNA热循环仪中在25℃培养10分钟,在42℃培养60分钟,然后在85℃培养5分钟。合成后,将cDNA储存在-20℃直至准备使用。预扩增:Taqman测试(LifeTechnologies)的预扩增池通过将10μL每种x20测试混合在一起而制备(共16种测试:4种感兴趣的基因(大鼠),4种管家基因(Housekeepers)(大鼠),4种感兴趣的基因(小鼠)和4种管家基因(小鼠))并稀释至x0.2(通过加入840μLTE缓冲液(LifeTechnologies))各25μLcDNA通过以下培养而扩增:试剂体积2xPreAmpMM25μL0.2X汇聚的测试混合物12.5μL使用以下循环条件:阶段重复温度时间酶活化(保持)1195℃10minsPCR(周期)21495℃30sec60℃4mins酶灭活99℃10mins将预扩增产物储存在-20℃直到准备使用,然后用0.1XTE(pH8.0)稀释1:10,然后在TaqManOpenArray上运行。实时定量PCR:根据LifeTechnologies指南设置并运行QuantStudio开放阵列。将2X实时PCR主混合物(LifeTechnologies;cat:4462164)混合,并组合以下组分:组分1个样品的体积2x开放阵列主混合物2.5μL水1.3μL将1.2μL每种预扩增的cDNA与3.8μL开放阵列主混合物混合,并置于Accufill384孔板中,通过AccufillTM系统分配到开放阵列上。OpenArray样品跟踪器软件用于跟踪样品。开放阵列在QuantStudio上运行,并在QuantStudio上进行分析和质量控制,并将生成的文件导出到ArrayStudio进行数据分析。通过QuantStudio开放阵列评估小鼠和大鼠坐骨神经中的CXCR2、CXCL1和NPY的表达:ArrayStudio中的分析:使用OpenArray条形码注释QuantStudio.txt文件,删除具有空目标名称值的所有行,并且将其适当排序为ArrayStudio的格式。单独的开放阵列.txt文件已上传到ArrayStudio中。检查“无逆转录”、“无模板对照”和“水空白”数据,并从最终分析中除去。对四种管家基因进行主成分分析(PCA)。在第一次PCA运行后去除异常值数据。在通过管家基因调整协方差的影响后,使用该模型估计每个基因和治疗的平均log2(丰度)。选择并运行全线性分析模型和分析因子。这产生了归一化数据的汇总表、去除了异常值的汇总表、每个基因的系数和指示基因是否通过归一化改进的P值。通过3因素主成分分析来分析这些数据,以检查生物复制样品聚集在一起。在每个模型和物种内通过单因素ANOVA方差分析来分析数据,以在每个时间点产生不同处理之间的倍数差值。计算平均差异不包含0的贝叶斯后验概率。取大于0.95的概率表示平均差异显著不同于0。对于CCI模型,比较类似组织的同侧和对侧样品。结果:当观察组织表达时,在涉及CCI的大鼠和小鼠实验之间的组间基线缩足阈值(PWT)没有差异。CCI诱导显著的机械性异常性疼痛表型,如通过大鼠(图1)和小鼠(图2)中PWT的显著减少所反映的。在大鼠中,与基线值和在这些时间点的假手术组相比,CCI组的PWT在手术后的第3天和第7天显著降低(图1)。(数值是平均值±SEM。**P<0.01,表示在相同时间点与假手术组相比的显著性,双因素ANOVA方差分析。基线:n=20/组;第3天:n=15/组;第7天:n=10/组)。类似地,小鼠在慢性压迫性损伤后发展机械性异常性疼痛。术后第3天和第7天,CCI组的PWT显著降低(图2)。(数值是平均值±SEM。***P<0.001,表示在相同时间点与假手术组相比的显著性,双因素ANOVA方差分析。第-1天:假手术组n=20,CCI组n=21;第0天:假手术组n=20,CCI组n=10;第3天:假手术组n=15,CCI组n=16;第7天:假手术组n=10,CCI组n=11)。从图3(大鼠)和图4(小鼠)可以看出,CXCR2和CXCL1在大鼠和小鼠中的表达谱在CCI诱导第1天后的坐骨神经中的CCI(同侧;ipsi)的位点处显著升高,如通过与假处理的动物相比的倍数变化的统计学显著增加所证明。当与假手术治疗的动物比较时,在CCI诱导后第3天,在坐骨神经中CCI(同侧;ipsi)的位点处的神经感觉肽(神经肽Y(NPY))的表达谱仅在大鼠中显著升高。从图3可以看出,与假手术组动物或对侧组织相比,CCI手术后的早期阶段,同侧坐骨神经中CXCR2及其配体CXCL1mRNA显著增加,在第7天减退。在第7天,与假手术动物和对侧组织相比,神经肽-Y(NPY)mRNA在统计学上不同。值显示为平均值。#表示在同一时间点CCI和假手术大鼠之间的显著性差异,*表示在相同时间点的对侧和同侧坐骨神经之间的显著性差异,在每个时间点n=5/组,通过贝叶斯后验概率>0.95评估。ipsi=同侧;contra=对侧。从图4(小鼠)可以看出,与假手术动物或对侧组织相比,CCI手术后的早期阶段,同侧坐骨神经中CXCR2及其配体CXCL1mRNA显著增加,在第7天减退。在进行测量的任何时间点,与假手术动物或对侧组织相比,神经肽-Y(NPY)mRNA没有统计学差异。值显示为平均值。#表示在同一时间点CCI和假手术小鼠之间的显著性差异,*表示在相同时间点对侧和同侧坐骨神经之间的显著性差异,在每个时间点n=5,通过贝叶斯后验概率>0.95评估。ipsi=同侧;contra=对侧。总结:在两种啮齿动物物种中的CCI程序引起稳健的机械性异常性疼痛。在这两种物种中,增强的CXCR2和CXCL1mRNA表达在机械性异常性疼痛的暂时评估的早期明显。在大鼠中感觉神经肽Y表达也有变化。大鼠中的这种特性与在CCI模型中CXCR2系统和感觉神经肽的已知灵敏度一致,并且允许该方法用于在奥沙利铂模型中检测这些蛋白质。h)在奥沙利铂-诱导的急性感觉损伤啮齿动物中的临床前基因表达将成年雄性Sprague-Dawley大鼠(230-300g)在恒定温度和湿度的受控环境(温度:21±1℃,光:暗周期为12:12小时)中以3~5只一组圈养在有机玻璃笼中,随意获得食物和水。将成年雄性C57BL6小鼠(21-30g)在恒定温度和湿度的受控环境(温度:21±1℃,光:暗周期为12:12小时)中以3~5只一组圈养在有机玻璃笼中,随意获得食物和水。允许动物从运输中恢复至少一周,然后开始实验。基线测试:所有动物在注射前进行机械性异常性疼痛的行为测试,以确定基线缩足阈值。在最后两天(第-1天和第0天)获取用von-Frey纤毛刺激的基线PWT。神经病变诱导:制备2mg/ml的奥沙利铂在5%葡萄糖中的溶液。对于大鼠,在第0天通过腹膜内途径以12mg/kg以6ml/kg的体积施用单剂量的奥沙利铂。载体对照组接受腹膜内6ml/kg的5%葡萄糖溶液。对于小鼠,在第0天通过腹膜内途径以15mg/kg以7.5ml/kg的体积施用单剂量的奥沙利铂。载体对照组接受腹膜内7.5ml/kg的5%葡萄糖溶液。行为部分:通过在施用单剂量的载体或奥沙利铂后测量PWT来确定机械性异常性疼痛的出现。在连续两天(第-1天和第0天)测定PWT,然后在第1天、第3天和/或第7天重新评估。大鼠:在试验前将动物置于凸起的金属网上的单独有机玻璃盒中至少40分钟。从最小力(约1g)的细丝开始,将每根细丝垂直地施加到爪的腹侧表面的中心,直到轻微弯曲6秒。如果动物在刺激时缩回或抬起爪子,则使用具有稍微低于测试的力的纤毛。如果没有观察到反应,则测试具有稍微更高的力的纤毛。将诱导可靠反应所需的最小量的力(5次试验中3次为阳性)记录为PWT的值。小鼠:在测试前将动物置于凸起的金属网上的单独的有机玻璃盒中30-40分钟。以1秒开1秒关的方案依次施加一系列渐变的vonFrey纤毛(0.07、0.16、0.4、0.6和1g),重复10次。将每根纤毛垂直地施加到爪的腹侧表面的中心,直到它略微弯曲。施加于动物后爪以在10次试验中诱导5次反应的力记录为PWT。组织取样:在实验结束时,用包含异氟烷+氧气的混合气体麻醉动物,并通过断头术杀死动物。手术切除来自每侧的坐骨神经的节段,在分析天平上称重并记录。将来自每侧的坐骨神经分别置于2mlCorning小瓶中。组织在液氮中快速冷冻,然后在-80度储存。统计分析:通过GraphpadPrism使用常规双因素ANOVA方差分析来分析行为数据,“治疗”作为受试者之间的效应,“天”作为受试者内的效应。使用Bonferroni方法进行事后分析。p值<0.05被认为是统计学显著的。基因表达部分:切除啮齿动物的坐骨神经并在液氮中快速冷冻。将组织在-80℃冷冻储存或在干冰上储存,直到处理用于CXCR2和CXCL1的RNA分析。ActB、GAPDH、HPRT1和MAPK6用作内部测试对照和标准品,并且NPY用作奥沙利铂诱导的损伤的对照。使用Q-PCR测量以下基因。测试用于制备预扩增池。大鼠的具体测试基因符号Taqman测试IDActBRn00667869_m1CXCL1Rn00578225_m1CXCR2Rn00567841_m1GAPDHRn01775763_g1HPRT1Rn01527840_m1MAPK6Rn00581152_m1NPYRn01410145_m1小鼠的具体测试基因符号Taqman测试IDActBMm01205647_g1CXCL1Mm04207460_m1CXCR2Mm00438258_m1GAPDHMm99999915_g1HPRT1Mm01545399_m1MAPK6Mm00727050_s1NPYMm03048253_m1总RNA分离:对于每个组织样品,根据制造商的说明,通过在RocheMagnaLyser上的GreenBead管(Roche)中的MagNAPureLC裂解缓冲液(Roche)中将组织匀浆制备一个RNA样品,并使用RocheMagnaPureRNA分离系统(Roche)根据制造商的说明进行总RNA分离。将样品以50μL等分试样洗脱,并在-80℃储存直至需要。RNA定量和完整性评估:根据制造商的说明,在LifeTechnologiesQubit上定量每个总RNA样品。使用AgilentPico试剂盒(AgilentTechnologies),根据制造商的说明,在Agilent2100Bioanalyser上评估总RNA样品的代表性样品的完整性。如果RIN为6或更多,则认为RNA是可接受的质量。第一链cDNA合成:使用SuperscriptVilo试剂盒(LifeTechnologies)从50ng的每种总RNA合成第一链cDNA。每个RNA用无RNase的水补足至11μL,并向每个RNA样品中加入14μL主混合物。主混合物基于下表制成:组分体积5xViloRT缓冲液5μL10xVilo酶混合物2.5μL无核酸酶的水6.5μL最终容积RTmastermix14μL将样品在DNA热循环仪中在25℃培养10分钟,在42℃培养60分钟,然后在85℃培养5分钟。合成后,将cDNA储存在-20℃直至准备使用。预扩增:Taqman测试(LifeTechnologies)的预扩增池通过将10μL每种x20测试混合在一起而制备[共16种测试:4种感兴趣的基因(大鼠),4种管家基因(大鼠)]并稀释至x0.2(通过加入840μLTE缓冲液(LifeTechnologies)]。各25μLcDNA等分试样通过以下培养而扩增:试剂体积2xPreAmpMM25μL0.2X汇聚的测试混合物12.5μL使用以下循环条件:阶段重复温度时间酶活化(保持)1195℃10minsPCR(周期)21495℃30sec60℃4mins酶灭活99℃10mins将预扩增产物储存在-20℃直到准备使用,且用0.1XTE(pH8.0)稀释1:10,然后在TaqManOpenArray上运行。实时定量PCR:根据LifeTechnologies指南设置并运行QuantStudio开放阵列。将2X实时PCR主混合物(LifeTechnologies;cat:4462164)混合,并组合以下组分:组分1个样品的体积2x开放阵列主混合物2.5μL水1.3μL将1.2μL每种预扩增的cDNA与3.8μL开放阵列主混合物混合,并置于Accufill384孔板中,通过AccufillTM系统分配到开放阵列上。OpenArray样品跟踪器软件用于跟踪样品。开放阵列在QuantStudio上运行,并在QuantStudio上进行分析和质量控制,并将生成的文件导出到ArrayStudio进行数据分析。通过QuantStudio开放阵列评估大鼠坐骨神经中的CXCR2、CXCL1、NPY和CGRP的表达。ArrayStudio中的分析:使用OpenArray条形码注释QuantStudio.txt文件,删除具有空目标名称值的所有行,并且将其适当排序为ArrayStudio的格式。单独的开放阵列.txt文件已上传到ArrayStudio中。检查“无逆转录”、“无模板对照”和“水空白”数据,并从最终分析中除去。对四种管家基因进行主成分分析(PCA)。在第一次PCA运行后去除异常值数据。在通过管家基因调整协方差的影响后,使用该模型估计每个基因和治疗的平均log2(丰度)。选择并运行全线性分析模型和分析因子。这产生了归一化数据的汇总表、去除了异常值的汇总表、每个基因的系数和指示基因是否通过归一化改进的P值。通过3因素主成分分析来分析这些数据,以检查生物复制样品聚集在一起。在每个模型和物种内通过单因素ANOVA方差分析来分析数据,以在每个时间点产生不同处理之间的倍数差值。计算平均差异不包含0的贝叶斯后验概率。计算平均差异不包含0的贝叶斯后验概率。如果概率大于0.95,则意味着平均差异显著不同于0。分别比较每个组织。结果:在涉及奥沙利铂的大鼠和小鼠实验之间,各组之间的基线PWT没有差异。从图5(大鼠)和图6(小鼠)可以看出,奥沙利铂治疗在两种啮齿动物物种中引起显著的机械性异常性疼痛,如从第1天维持至第7天的PWT的显著降低所表示的。从图5可以看出,大鼠在单剂量的奥沙利铂后产生机械性异常性疼痛。在第1、3和7天,奥沙利铂组的PWT统计学下降。值为平均值±SEM。*P<0.05,***P<0.001,表示与载体组相比在相同时间点的显著性,2因素ANOVA方差分析。第-2天、第-1天、第0天:n=20/组;第1天:n=5/组;第3天:n=15/组;第7天:n=10/组。从图6中可以看出,小鼠在单次剂量的奥沙利铂后产生机械性异常性疼痛。在第1、3和7天,奥沙利铂组的PWT统计学下降。值为平均值±SEM。*P<0.05,**P<0.01,***P<0.001,表示与载体组相比在相同时间点的显著性,双因素ANOVA方差分析。第-2天,第-1天,第0天:n=20/组;第1天:n=5/组;第3天:n=15/组;第7天:n=10/组。从图7(大鼠)和图8(小鼠)可以看出,在奥沙利铂治疗后第1天,在大鼠左坐骨神经中,奥沙利铂治疗后CXCR2和NPYmRNA的表达谱显著升高,但在小鼠中不明显。在两种物种中在奥沙利铂治疗后,左侧坐骨神经中的奥沙利铂治疗后CXCL1mRNA的表达谱没有统计学差异。从图7可以看出,在左侧坐骨神经中,CXCR2及其配体CXCL1mRNA在注射奥沙利铂后的早期(第1天)高度增加,如同NPYmRNA。值显示为平均值。#表示在同一时间点奥沙利铂和载体组之间的显著性差异,在每个时间点n=10,通过贝叶斯后验概率>0.95评估。从图8可以看出,在左侧坐骨神经中,在奥沙利铂注射后7天内,CXCR2或其配体CXCL1mRNA和NPYmRNA都不显著增加。值显示为平均值。#表示在同一时间点奥沙利铂和载体组之间的显著性差异,在每个时间点n=10,通过贝叶斯后验概率>0.95评估。总结:在单次奥沙利铂给药后,大鼠和小鼠都出现出稳健的机械性异常性疼痛。然而,CXCR2和NPYmRNA表达(在左坐骨神经中增加)的早期统计学显著变化仅在大鼠中明显。因此,选择大鼠作为主要物种,其能够探索药理学抑制CXCR2受体对奥沙利铂诱导的机械性异常性疼痛的作用。i)临床前CXCR2拮抗剂对奥沙利铂诱导的急性感觉损伤大鼠的作用动物:将60只成年雄性Sprague-Dawley大鼠(CharlesRiver,UK。230-300g)用于本研究。将动物在恒定温度和湿度(温度:21±1℃,光:暗周期为12:12小时)的受控环境中以3至5只每组饲养在有机玻璃笼中,随意获得食物和水。在开始实验前,允许它们从运输中恢复至少一周。通过湿磨法制备式(A)化合物的0.5mg/ml、2mg/ml和5mg/ml混悬液(分别针对5mg/kg,20mg/kg和50mg/kg给药),然后在1%甲基纤维素(MC)中超声处理。每周制备制剂,并在避光保存于4℃。给药体积为10ml/kg。在整个给药期间连续搅拌化合物。在水中制备1%(1g/100ml水)的甲基纤维素(Sigma),并置于搅拌器上直至完全溶解。载体处理的动物以每日两次(b.i.d.)给药1%MC,10ml/kgp.o。通过在5%葡萄糖(Baxter)中超声处理制备作为2mg/ml溶液(针对12mg/kg)的奥沙利铂(Tocrisbioscience)。制剂在给药当天新鲜制备。给药体积为6ml/kg。研究方案:研究方案示于表1中。在第0天注射一次奥沙利铂,从第-1天至第6天每日两次给予三个剂量的式(A)化合物。在第-2天、第-1天、第0天、第3天和第7天测试PWT。表1研究以下5组:诱导化疗诱发的神经病:以6ml/kg的体积以12mg/kg腹膜内(i.p.)注射单剂量的奥沙利铂。在使用前将奥沙利铂溶于5%葡萄糖中至2mg/ml。使用施加到后爪上的一系列渐变的vonFrey纤毛以引发缩足反应(PWT,参见下文)来监测神经性疼痛的出现(其特征在于显著的机械性异常性疼痛),。载体对照组接受腹膜内6ml/kg的5%葡萄糖溶液。以相同的测试顺序注射动物。在试验前将动物置于凸起的金属网上的单独有机玻璃盒中至少40分钟。从最小力(约1g)的细丝开始,将每根细丝垂直地施加到爪的腹侧表面的中心,直到轻微弯曲,保持6秒。如果动物在刺激时缩回或抬起爪子,则使用具有稍稍低于测试的力的纤毛。如果没有观察到反应,则测试具有稍微更高的力的纤毛。将诱导可靠反应所需的最小量的力(5次试验中3次为阳性)记录为PWT的值。在连续三天(第-2天,第-1天和第0天)评估PWT,并在施用单剂量的载体或奥沙利铂之后在第3天和第7天重新评估,以监测机械性异常性疼痛的出现。第-2天和第-1天是在给予奥沙利铂之前的基线,第0天是在奥沙利铂注射当天的读数。统计分析:通过双因素重复量数ANOVA方差分析来分析行为数据,“治疗”作为受试者之间的效应,“天”作为受试者内的效应。事后分析使用计划的成对比较进行[InVivoStat;Clark等人,J.Psychopharmacology,26(8)1136-1142(2012)]。结果:注射奥沙利铂导致机械性异常性疼痛的出现,其使用PWT的von-Frey评估测量。在奥沙利铂之前用式(A)化合物(在第-1天开始)的预防性治疗导致预防奥沙利铂诱导的机械性异常性疼痛的出现。这在奥沙利铂治疗后第3天的所有剂量水平和在奥沙利铂治疗后第7天的20和50mg/kgbid的情况下是明显的。如图9所示,奥沙利铂的推注导致明显的机械性异常性疼痛的出现。在第3天和第7天,与Veh/Veh组相比,PWT显著降低。预防性的式(A)化合物给药抑制了通过奥沙利铂治疗建立的机械性异常性疼痛,PWT与Oxa/Veh组相比显著增加。数值为平均值±SEM。*P<0.05,**P<0.01和***P<0.001指示与Oxa/Veh组相比的显著性,++p<0.01和+++p<0.001指示与Veh/Veh组相比的显著性,2因素ANOVA方差分析。Oxa=奥沙利铂;Veh=载体;5、20和50mpk=式(A)化合物的剂量水平,以mg/kgbidpo。总结:单次推注奥沙利铂导致大鼠机械性异常性疼痛的出现。用式(A)化合物的预防性治疗减轻了机械性异常性疼痛的出现。在第3天,与奥沙利铂和载体组相比,式(A)化合物的所有三个剂量水平(5、20和50mg/kgp.o.b.i.d)都降低了机械异常性疼痛。到第7天,仅在以20和50mg/kgp.o.b.i.d接受式(A)化合物的动物中仍然存在机械性异常性疼痛的稳健降低。j)临床前CXCR2拮抗剂对重复给药奥沙利铂诱导的感觉损伤大鼠的作用已经确认化疗治疗引起人和啮齿类动物中神经的传导潜能的显著降低[Renn等人,Pain,26;7:29,(2011)]。因此,为了补充研究奥沙利铂诱导的行为超敏性,进行坐骨神经的电生理研究以进一步探测CXCR2机理参与人类经历的化疗诱导的周围神经病变的神经生理学性质。将雄性Sprague-Dawley大鼠(109-167g,周龄)在恒定温度和湿度(温度:25±3℃,光:暗周期为12:12小时)的控制环境中以3只一组圈养在笼中,随意获得食物(25g/大鼠/天的LabDiet5053)和水。化合物制备:将式(A)化合物制备为在1%甲基纤维素(MC)(w/v)中的2mg/mL悬浮液。在水中制备1%(w/v)(1g/100mL水)的MC(Sigma目录号423238),并置于搅拌器上直至完全溶解。该制剂每6-8天制备一次,每次400-700mL,并在4℃避光储存。所用的给药体积为10mL/kg,得到20mg/kgp.o.剂量。载体处理的动物以10mL/kgp.o每日两次(b.i.d.)给药1%MCw/v。将奥沙利铂(Selleckchem)加入5%葡萄糖(SigmaAldrich)溶液中,超声处理30分钟直至溶解。在每次注射前制备新鲜溶液。对照载体动物用5%w/v葡萄糖以5ml/kg处理。在milli-Q水中制备5%(w/v)(5g/100mL水)的葡萄糖(SigmaAldrichCat#G7021),并置于搅拌器上直至完全溶解。使用无菌0.2μm过滤器(ThermoscientificCat#595-4520)过滤,并在室温储存。用SpragueDawley大鼠的治疗方案:对雄性SpragueDawley大鼠每天用式(A)化合物(20mg/kgp.o.b.i.d.)或其载体(1%甲基纤维素10ml/kgp.o.b.i.d.)给药4周(28天),开始于第0天。每天早晨进行一次给药,随后约8小时后在晚上进行第二次给药。在第一天用式(A)化合物预处理后二十四小时,在短暂的3%异氟烷(Abbott)麻醉下给动物施用奥沙利铂(4mg/kgi.p.)或其载体(葡萄糖溶液5%w/v:1ml/kgi.p.)。在早晨用式(A)化合物或载体预处理90分钟后,用奥沙利铂或其载体的这种治疗每周重复两次持续4周(在第1、3、8、10、15、17、22、24天)。因此,将大鼠随机分配到以下组:体重跟踪:在第1、3、9、11、16、18、23和25天,每周两次测量大鼠的体重,持续4周。第5周的体重值表示当进行传导速度研究时从第29、30和31天的每个组的组合数据。进行这些研究的科学家不知道动物接受的治疗,直到所有数据分析完成。使用GraphpadPrism软件分析数据。它们以平均值±SEM表示。通过双因素ANOVA方差分析,随后通过Dunnett检验测定参数差异的统计显著性。p值<0.05被认为是统计学显著的。坐骨神经传导速度的测量:在用式(A)的化合物或其载体治疗4周后,在第29天至第31天之间在右坐骨神经中测量神经传导速度。在这些天的每一天,平衡治疗组,进行这些研究的科学家不知道动物接受的治疗,直到所有数据分析完成。在测量传导速度之后,收集大鼠的左坐骨神经用于组织学研究(参见下面的方法)。用水合氯醛(350mg/kg,3.5mL/kg,腹膜内)麻醉大鼠。当达到麻醉的手术深度时,小心暴露坐骨切口上方的肌肉和右后肢踝部。仔细鉴定右坐骨神经,并通过放置在第二和第三手指之间的电极记录动作电位,如Jamieson等人,Br.J.Cancer,88(12):1942-7(2003)所述,以及在坐骨切口和踝处经由铂丝电极电刺激神经(5V,0.5s,单波脉冲),来测量传导速度。测量坐骨切口和踝之间的长度。使用具有CED1902mkIV可编程放大器/滤波器和DS2A-MK.II单独刺激器-恒定电压(CambridgeElectronicDesignLimited)的CEDMicro1401-3科学数字数据记录器记录动作电位,数据通过CEDSignal(Windows软件版本6)(CambridgeElectronicDesignLimited)分析。神经传导速度被计算为坐骨切口和踝之间的长度除以各刺激位点处的时间潜伏期之间的差异[潜伏期(坐骨切口)–潜伏期(踝)]。使用GraphpadPrism软件分析数据。它们以平均值±SEM表示。通过单因素ANOVA,随后通过Tukey检验测定参数差异的统计学显著性。p值<0.05被认为是统计学显著的。坐骨神经的组织学评估:在传导速度的测量之后,收集大鼠的左坐骨神经用于组织学研究。进行这些研究的科学家不知道动物接受的治疗,直到所有数据分析完成。定位并小心地切除左坐骨神经(远端)的1cm部分。将神经在4%多聚甲醛(SigmaAldrich目录号:P6148)中固定过夜,然后用0.2%甘氨酸(SigmaAldrich目录号:15527)固定过夜。用磷酸盐缓冲盐水[磷酸二氢钠(SigmaAldrich目录号:04270)];磷酸氢二钠(SigmaAldrich目录号:30427);氯化钠(BDH目录号:7647-14-5)]洗涤后,将坐骨神经在2%四氧化锇中后固定2小时。在从30%至70%乙醇脱水后,将神经样品包埋在石蜡中,将神经切成2μm厚度的切片(使用Leica切片机),安装在载玻片(SuperiorMaerienfeld)上,并在58℃风干。然后将切片浸入100%二甲苯(BDH;CAS号1330-20-7)中,随后用100%至70%乙醇(Merck;CAS号:64-17-5),然后用100%Milli-Q水再水合。然后将神经在1%甲苯胺蓝中染色,并在用水和75%乙醇洗涤后,用盖玻片固定载玻片。选择第一清晰和完整的神经部分用于分析。使用自动立式复合显微镜(LeicaDM6000B)捕获图像。使用INCellAnalyzer6000(GEHealthcare)自动测量坐骨神经(包括髓鞘)的神经纤维的内部的横截面积和整体面积,如DiCesareMannelli等人,J.Pain,14(12):1585-600(2013)中所述。然后计算坐骨神经的神经纤维的内部面积与整体面积的比率作为神经的髓鞘的厚度的指示。使用GraphpadPrism软件分析数据。它们以平均值±SEM表示。通过单因素ANOVA方差分析,随后通过Tukey检验测定参数差异的统计学显著性。p值<0.05被认为是统计学显著的。结果:使用慢性大鼠CIPN模型,检查体重、坐骨神经髓鞘的传导速度和厚度。在第29天、第30天和第31天测量神经传导速度。在第29、30或31天之间的每个治疗组中未观察到传导速度的差异。相比载体-处理的对照(40.28±1.30m/s),奥沙利铂治疗的大鼠显示出在坐骨切口和脚踝之间显著更低的传导速度(35.99±0.59m/s)(p<0.05)。式(A)化合物治疗显著地防止大鼠中奥沙利铂诱导的传导速度降低(40.83±1.45m/s)(p<0.05)(图10)。在载体/奥沙利铂处理的和式(A)的化合物/奥沙利铂处理的组之间没有观察到体重的显著性差异(图11)。使用组织学染色研究式(A)化合物对坐骨神经髓鞘厚度的影响。来自所有组的神经纤维的代表性横截面显示在图12中。对神经的髓鞘的厚度进行定量。从图13,与载体对照(0.13±0.01)相比,经奥沙利铂处理的大鼠显示出髓鞘的显著变薄(由更高的神经纤维的内部面积/整体面积的比例指示(0.23±0.02))(p<0.001)。与载体/奥沙利铂处理相比,向经奥沙利铂处理的大鼠口服施用式(A)化合物显示出坐骨神经的髓鞘厚度增加,如通过较低的神经纤维的内部面积/整体面积比例指示(0.17±0.01)(p<0.05)。因此,用式(A)化合物的预防性治疗抑制了由奥沙利铂治疗诱导的坐骨神经传导速度受损。图10:值是平均值±SEM。*P<0.05表示载体/奥沙利铂与载体/载体组相比的显著性,#p<0.05表示式(A)的化合物/奥沙利铂与载体/奥沙利铂组相比的显著性,单因素ANOVA方差分析。载体/载体,n=13;载体/奥沙利铂,n=12;式(A)的化合物/奥沙利铂,n=12。在图11中显示,式(A)的化合物不影响奥沙利铂诱导的体重变化。在2周时间点,在载体/载体和载体/奥沙利铂治疗组之间观察到体重的显著性差异。每周两次测量体重,持续4周。第5周的体重值表示当进行传导速度研究时从第29、30和31天的每个组的组合数据。在研究的任何时间点,载体/奥沙利铂和式(A)化合物/奥沙利铂治疗组之间没有发现显著性差异。数值为平均值±SEM。***p<0.001指示与载体/奥沙利铂相比载体/载体的显著性,###p<0.001指示与化合物D/奥沙利铂处理的大鼠相比载体/载体的显著性;双因素ANOVA方差分析,然后是Dunnett检验,n=13(载体/载体),n=12(载体/奥沙利铂),n=12(化合物D/奥沙利铂)。图12和13显示式(A)的化合物防止奥沙利铂使坐骨神经的髓鞘厚度减小。图12示出了使用光学显微镜在40x放大率下拍摄的来自每组的图像。图13图示了神经纤维的髓鞘的厚度。评价内神经纤维的横截面积与坐骨神经整个神经纤维(包括髓鞘)的总横截面积的比率。奥沙利铂减少大鼠坐骨神经的髓鞘的厚度,通过用式(A)的化合物处理逆转该效应。数值为平均值±SEM。***p<0.001指示与载体/载体组相比的载体/奥沙利铂的显著性,#p<0.05指示与载体/奥沙利铂处理的大鼠相比的化合物D/奥沙利铂的显著性;单因素ANOVA方差分析随后是Tukey检验,n=12(载体/载体),n=12(载体/奥沙利铂),n=12(式(A)的化合物/奥沙利铂)。总结:奥沙利铂诱导大鼠的周围神经病变,通过神经传导速度和神经髓鞘的厚度的降低评估。用式(A)化合物的预处理防止了奥沙利铂治疗的大鼠中坐骨神经传导速度的降低,但不影响奥沙利铂治疗诱导的体重减少。式(A)的化合物还改善由奥沙利铂诱导的坐骨神经的髓鞘的变化。这些研究的结果表明,式(A)化合物防止在大鼠中奥沙利铂诱导的行为特征的变化(该行为特征为对机械刺激的超敏反应)、坐骨神经的髓鞘状态和神经传导速度的减慢。这些是患者中化疗诱导的周围神经病变的特征。k)机理临床研究的证明打算进行机理临床研究的证明以进一步证明式(A)化合物在治疗人类患者的CIPN中的作用。可能的研究设计如下:临床研究设计将是:双盲、安慰剂对照的平行组,机理研究证据在患有结肠直肠癌的患者中,计划由含奥沙利铂的化疗治疗开始。该项目的概况可能包括受试者每两周出席抗癌化疗的临床中心。在每次奥沙利铂给药之前,将根据临床中心程序进行常规实验室检查。指示受试者在奥沙利铂iv输注前两天开始服用式(A)化合物。在前六次奥沙利铂输注期间,将指导合格的受试者每天一次在早晨服用一剂的式(A)化合物(x片剂xmg或安慰剂),持续7天。设计式(A)化合物的给药方案以在奥沙利铂的全身峰值期间实现CXCR2拮抗剂最大效应。在该第一研究中,间歇给药将通过延长与奥沙利铂的共同给药来最大化治疗功效并最小化任何副作用的可能性。在第六周期结束后,受试者将停止用式(A)化合物或安慰剂的治疗,并将继续临床上合适的化疗方案。在前7个化疗周期期间,在临床中心时,评估将包括安全性、耐受性和药代动力学。在基线和在第1、3、5、6和7周期的奥沙利铂输注后24-48小时之间进行关于神经兴奋性概况的临床评估和主要电生理学测量。对临床中心的所有随访将包括使用NCI-CTCv4和总神经病评分和亚组[TNSc;CavalettiG.F.B.,J.Peripher.Nerv.Syst.,Sep.,12(3),210-5(2007);CornblathDR,Neurology,53(8),1660(1999)]的临床评估。将根据护理标准监测肿瘤学措施。主要措施包括:·测量的感觉兴奋性(超兴奋性%)的变化,其作为化疗周期1的给药前和在最后的式(A)的化合物/安慰剂给药后的化疗周期前的差异。式(A)化合物的安全性:监测不良事件、临床体征、安全实验室和生命体征。·式(A)的化合物的PK。式(D)化合物:1-(4-氯-3-((1,4-二甲基哌啶-4-基)磺酰基)-2-羟基苯基)-3-(2-氯-3-氟苯基)脲三氟乙酸盐向6-氨基-3-氯-2-[(1,4-二甲基-4-哌啶基)磺酰基]苯酚二盐酸盐(中间体D1)(0.30g)在1,4-二噁烷(10mL)和水(1mL)中的溶液中添加NaHCO3(0.193g)。将混合物在室温搅拌10分钟。然后添加2-氯-1-氟-3-异氰酸酯基苯(0.131g)。搅拌再持续10分钟。然后,该反应混合物用水(50mL)稀释,用EA(2×50mL)萃取。该溶液然后用盐水洗涤,用无水Na2SO4干燥且通过MDAP纯化以得到标题产物(130mg)。中间体D1:6-氨基-3-氯-2-((1,4-二甲基哌啶-4-基)磺酰基)苯酚二盐酸盐步骤1:在-78℃向4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)哌啶-1-甲酸叔丁酯(0.35g)(中间体D2)在四氢呋喃(THF)(25mL)中的溶液中添加正丁基锂(0.383mL,2.0M在环己烷中)。将混合物在-78℃搅拌1h。然后添加MeI(0.048mL)。搅拌在-78℃持续4h。然后,该反应用NH4Cl水溶液淬灭,用EA萃取(2×50mL)。该溶液然后用盐水洗涤,用无水Na2SO4干燥,且浓缩以得到4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)-4-甲基哌啶-1-甲酸叔丁酯(0.4g)。步骤2:向4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)-4-甲基哌啶-1-甲酸叔丁酯(0.35g)在二氯甲烷(DCM)(20mL)中的溶液中添加TFA(0.572mL)。将混合物在室温搅拌过夜。所得溶液真空浓缩以得到2-(叔丁基)-6-氯-7-((4-甲基哌啶-4-基)磺酰基)苯并[d]噁唑三氟乙酸盐(0.35g)。步骤3:向2-(叔丁基)-6-氯-7-((4-甲基哌啶-4-基)磺酰基)苯并[d]噁唑三氟乙酸盐(0.35g)在N,N-二甲基甲酰胺(DMF)(10mL)中的溶液中添加AcOH(0.054mL)和甲醛(0.788mL)。然后将反应混合物冷却至0℃且搅拌10分钟。然后分批添加三乙酰氧基硼氢化钠(0.6g)。反应完成后,将混合物用饱和NaHCO3(20mL)水溶液淬灭且用EA萃取(2×50mL)。该溶液然后用盐水洗涤,用无水Na2SO4干燥,且浓缩以得到2-(叔丁基)-6-氯-7-((1,4-二甲基哌啶-4-基)磺酰基)苯并[d]噁唑(0.3g)。步骤4:向2-(叔丁基)-6-氯-7-((1,4-二甲基哌啶-4-基)磺酰基)苯并[d]噁唑(0.3g)在1,4-二噁烷(10mL)和水(10mL)中的溶液中添加HCl(0.640mL,37%在水中)。将混合物在120℃加热3小时。然后,所得溶液真空浓缩以得到标题产物(0.3g),将其用于下一步而不用进一步纯化。中间体D2:4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)哌啶-1-甲酸叔丁酯步骤1:向4-羟基哌啶-1-甲酸叔丁酯(10.0g)在DCM(100mL)中的溶液中添加TEA(13.9mL),然后在冰浴中添加MsCl(4.7mL)。将混合物在室温搅拌4小时。添加水(100mL)。分离有机层,用硫酸钠干燥,过滤且浓缩以得到4-((甲基磺酰基)氧基)哌啶-1-甲酸叔丁酯(13.9g),其为白色固体。MS(ES+)m/z302(MNa+)。步骤2:向2-(叔丁基)-6-氯苯并[d]噁唑-7硫醇钠(12.0g)和4-((甲基磺酰基)氧基)哌啶-1-甲酸叔丁酯(13.9g)在DMF(100mL)中的溶液中添加碳酸钾(6.3g)。将混合物在80℃搅拌2小时。添加EA(200mL)。有机相用盐水洗涤(4×200mL)。合并的有机层用硫酸钠干燥,过滤且浓缩以得到4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)硫基)哌啶-1-甲酸叔丁酯(19.3g),其为黄色油状物。MS(ES+)m/z369(M-t-Bu+H+H+)。步骤3:在0℃向4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)硫基)哌啶-1-甲酸叔丁酯(19.3g)在DCM(50mL)中的溶液中添加mCPBA(20.4g)。在室温搅拌过夜后,将混合物用NaHCO3水溶液和Na2S2O3水溶液淬灭,然后用EA萃取(2×150mL)。将合并的有机层洗涤,干燥且浓缩。粗物质通过在甲醇/H2O中重结晶纯化以得到4-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)哌啶-1-甲酸叔丁酯(14.0g)。MS(ES+)m/z479(MNa+)。式(E)化合物:(R)-1-(4-氯-2-羟基-3-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯基)-3-(2-氯环戊-2-烯-1-基)脲向6-氨基-3-氯-2-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯酚盐酸盐(50mg)(中间体E1)在吡啶(5mL)中的溶液中添加新鲜的(R)-1-氯-5-异氰酸酯基环戊-1-烯(中间体E2,0.03M在甲苯中,5mL)且所得混合物在室温搅拌过夜。将混合物用乙醇(5mL)淬灭且在减压下浓缩。残余物用MDAP纯化(碱性条件)以得到标题化合物(34mg);1H-NMR(400MHz,DMSO-d6)δppm10.36(br.s.,1H),8.39(d,J=8.8Hz,1H),8.20(s,1H),7.35(d,J=8.8Hz,1H),7.13(d,J=9.0Hz,1H),5.96-6.02(m,1H),4.69-4.80(m,1H),3.84(dd,J=11.7,4.4Hz,2H),3.49(t,J=11.2Hz,2H),2.22-2.46(m,3H),2.04(td,J=12.5,5.1Hz,2H),1.61-1.74(m,1H),1.58-1.61(m,1H),1.54-1.58(m,1H),1.50(s,3H);MS(ES+)m/z449(MH+)。中间体E1:6-氨基-3-氯-2-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯酚盐酸盐步骤1:向四氢-2H-吡喃-4-醇(10.0g)在DCM(200mL)中的溶液中添加TEA(12.9g)和甲烷磺酰氯(11.3g)。将混合物在0℃搅拌1小时,然后用H2O洗涤。有机层用Na2SO4干燥且浓缩以得到四氢-2H-吡喃-4-基甲磺酸酯(15.5g)。步骤2:向2-(叔丁基)-6-氯苯并[d]噁唑-7-硫醇(18.0g)和Cs2CO3(12.1g)在乙腈(5mL)中的溶液中添加四氢-2H-吡喃-4-基甲磺酸酯(14.8g)。将混合物在90℃搅拌16小时。冷却至室温后,将混合物浓缩。残余物在EA(100mL)和H2O(100mL)之间分配。将有机层干燥且浓缩以得到2-(叔丁基)-6-氯-7-((四氢-2H-吡喃-4-基)硫基)苯并[d]噁唑(24.0g)。MS(ES+)m/z326(MH+)。步骤3:向2-(叔丁基)-6-氯-7-((四氢-2H-吡喃-4-基)硫基)苯并[d]噁唑(24.0g)在DCM(1000mL)中的溶液中添加mCPBA(31.8g)。将混合物在15℃搅拌2小时,然后用Na2SO3水溶液淬灭。将pH调节至~7。将有机层干燥且浓缩以得到2-(叔丁基)-6-氯-7-((四氢-2H-吡喃-4-基)磺酰基)苯并[d]噁唑(18.0g)。步骤4:在-78℃在氮气氛向2-(叔丁基)-6-氯-7-((四氢-2H-吡喃-4-基)磺酰基)苯并[d]噁唑(5.0g)在THF(50mL)中的溶液中添加BuLi(2.5M在己烷中,6.2mL)。将混合物在-78℃搅拌45分钟。添加MeI(2.2g)。将反应混合物在-78℃搅拌1小时,然后用NH4Cl水溶液淬灭。将有机层干燥且浓缩。残余物通过柱色谱法纯化以得到2-(叔丁基)-6-氯-7-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯并[d]噁唑(4.8g)。MS(ES+)m/z372(MH+)。步骤5:向2-(叔丁基)-6-氯-7-((4-甲基四氢-2H-吡喃-4-基)磺酰基)苯并[d]噁唑(1.0g)在1,4-二噁烷(10mL)中的溶液中添加HCl水溶液(37%,10mL)。在110℃回流4小时后,将混合物浓缩以得到标题化合物(1.0g),其为灰色固体。MS(ES+)m/z306(MH+)。中间体E2:(R)-1-氯-5-异氰酸酯基环戊-1-烯步骤1:向环戊-2-烯酮(1.2g)在甲醇(10mL)中的溶液中添加过氧化氢溶液(30%,0.5g)。所得混合物在室温搅拌过夜。添加冷却水(30mL)且所得混合物用饱和NaHCO3溶液中和。水层用DCM萃取(2×100mL)。合并的有机层用Na2SO4干燥,过滤且真空浓缩以得到6-氧杂双环[3.1.0]己-2-酮(1.3g),其为黄色油状物。步骤2:向6-氧杂双环[3.1.0]己-2-酮(25g)在甲醇(10mL)和水(3mL)中的溶液中添加氯化铈(III)七水合物(95g)。所得混合物在70℃搅拌1小时。添加冷却水(30mL)且所得混合物用饱和NaHCO3溶液中和。水层用DCM萃取(2×100mL)。合并的有机层用Na2SO4干燥,过滤且真空浓缩以得到2-氯环戊-2-烯酮(29g),其为黄色油状物。步骤3:向2-氯环戊-2-烯酮(600mg)在甲醇(20mL)中的溶液中添加氯化铈(III)七水合物(1918mg)和NaBH4(195mg)。所得混合物在室温搅拌1小时。添加冷水(30mL)且所得混合物用饱和NaHCO3溶液中和。水层用DCM萃取(2×100mL)。合并的有机层用Na2SO4干燥,过滤且真空浓缩以得到2-氯环戊-2-烯醇(400mg),其为黄色油状物。步骤4:在0℃向2-氯环戊-2-烯醇(20.0g)和异二氢吲哚-1,3-二酮(37.2g)在THF(200mL)中的溶液中添加Ph3P(66.4g)和DIAD(49.2mL)。将混合物在室温搅拌过夜。将溶剂真空去除且残余物通过柱色谱法纯化(用PE:EA=10:1洗脱)以得到2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(18.0g),其为黄色固体。MS(ES+)m/z248(MH+)。步骤5:2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(14g)通过SFC纯化以得到(R)-2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(5.0g),其为白色固体,和(S)-2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(5.5g),其为黄色油状物。(R)-2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮:手性HPLC(柱:AD-H(250*4.6mm,5um);流动相:MeOH/CO2=15%;流速:3.0ml/min;温度:40℃):tR=2.54分钟,ee%=100%;1H-NMR(400MHz,CDCl3)δppm7.74-7.98(m,4H),6.04(d,J=2.2Hz,1H),5.32(dd,J=9.3,1.9Hz,1H),2.77(ddd,J=9.4,8.0,3.0Hz,1H),2.41-2.62(m,2H),2.22-2.39(m,1H);(S)-2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮:手性HPLC(柱:AD-H(250*4.6mm,5um);流动相:MeOH/CO2=15%;流速:3.0ml/min;温度:40℃):tR=3.04分钟,ee%=100%;1H-NMR(400MHz,CDCl3)δppm7.32-8.36(m,4H),6.03(d,J=2.2Hz,1H),5.31(dd,J=9.3,1.8Hz,1H),2.77(ddd,J=9.4,8.0,3.0Hz,1H),2.40-2.64(m,2H),2.32(ddd,J=14.2,8.7,3.8Hz,1H)。步骤6:向(R)-2-(2-氯环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(3.5g)在乙醇(100mL)中的溶液中添加肼.H2O(0.7mL)。回流3小时后,将反应混合物冷却至室温。将沉淀过滤且用EtOH(10mL)冲洗。将滤液浓缩以去除一半溶剂。向溶液添加醚中的HCl(1M,20mL)且浓缩以得到(R)-2-氯环戊-2-烯胺的盐酸盐(2.0g)。1H-NMR(400MHz,DMSO-d6)δppm8.53(s,3H),6.22(s,1H),4.19(d,J=5.8Hz,1H),2.49-2.54(m,1H),2.26-2.45(m,2H),1.90-2.04(m,1H)。步骤7:向(R)-2-氯环戊-2-烯胺盐酸盐(600mg)在甲苯(15mL)中的溶液中添加碳酸二(三氯甲基)酯(694mg)。将混合物在120℃搅拌4小时。混合物然后冷却至室温以得到(R)-1-氯-5-异氰酸酯基环戊-1-烯的甲苯溶液。该溶液应每次新鲜合成。式(F)的化合物:(R)-1-(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲向4-氨基-2-(叔丁基磺酰基)-3-羟基苄腈(中间体F1,450mg)在吡啶(20mL)中的溶液中添加新鲜的在甲苯(20mL)中的(R)-5-异氰酸酯基-1-甲基环戊-1-烯(中间体F2,327mg)。将反应混合物在室温搅拌过夜。将混合物用水(5mL)淬灭且浓缩。残余物用MDAP纯化(酸性条件)以得到(R)-1-(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲(120mg)。1H-NMR(400MHz,DMSO-d6)δppm10.13(br.s.,1H),8.51-8.64(m,2H),7.60(d,J=8.6Hz,1H),7.34(d,J=8.3Hz,1H),5.52(s,1H),4.54(d,J=6.8Hz,1H),2.11-2.37(m,3H),1.66(s,3H),1.46-1.60(m,1H),1.30-1.46(m,9H);MS(ES+)m/z378(MH+)。中间体F1:4-氨基-2-(叔丁基磺酰基)-3-羟基苄腈步骤1:该反应以五批次进行(各600mg)微波合成,然后组合纯化:将2-(叔丁基)-7-(叔丁基磺酰基)-6-氯苯并[d]噁唑(中间体F3,0.6g)和氰化铜(I)(1.6g)在NMP(4mL)中的混合物在180℃在微波中搅拌90分钟。冷却后,合并5个批次,且用EA(100mL)和水(100mL)稀释。过滤后,分离有机层,洗涤,干燥,过滤且浓缩。残余物通过柱色谱法纯化(用PE:EA=1:0至7:3洗脱)以得到2-(叔丁基)-7-(叔丁基磺酰基)苯并[d]噁唑-6-甲腈(1.0g)。MS(ES+)m/z321(MH+)。步骤2:向2-(叔丁基)-7-(叔丁基磺酰基)苯并[d]噁唑-6-甲腈(1.0g)在乙醇(25mL)和水(25mL)中的溶液中添加氢氧化钠(0.3g)。所得混合物在60℃搅拌1小时,然后在减压下浓缩。残余物用水(50mL)稀释,用柠檬酸水溶液酸化至pH=6,且用EA萃取(2×50mL)。将合并的有机层洗涤,干燥,过滤且浓缩以得到N-(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)特戊酰胺(1.1g)。MS(ES+)m/z361(MH+)。步骤3:向N-(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)特戊酰胺(1.2g)在THF(30mL)中的溶液中添加DMAP(0.04g)和Boc2O(1.6mL)。将混合物在60℃搅拌2小时。向混合物添加肼.H2O(1.6mL)。所得混合物在室温搅拌过夜,用水(50mL)稀释,且用EA萃取(2×100mL)。将合并的有机层洗涤,干燥,过滤且浓缩。残余物通过柱色谱法纯化(用PE:EA=1:0至7:3洗脱)以得到(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)氨基甲酸叔丁酯(1.2g)。MS(ES+)m/z355(MH+)。步骤4:向(3-(叔丁基磺酰基)-4-氰基-2-羟基苯基)氨基甲酸叔丁酯(1.2g)在DCM(25mL)中的溶液中添加TFA(2.6mL)。所得混合物在室温搅拌过夜。去除DCM。残余物用NaHCO3水溶液(pH=8)碱化,且用EA萃取(2×50mL)。将合并的有机层洗涤,干燥,过滤且浓缩以得到标题化合物(0.8g)。MS(ES+)m/z255(MH+)。中间体F2:(R)-5-异氰酸酯基-1-甲基环戊-1-烯步骤1:将2-甲基环戊烷-1,3-二酮(27.0g)、2-甲基丙-1-醇(62.5g)和TsOH(4.6g)在苯(500mL)中的溶液加热至回流过夜。将溶剂真空去除且残余物真空蒸馏以得到3-异丁氧基-2-甲基环戊-2-烯酮(34.5g),其为黄色油状物。1H-NMR(400MHz,CDCl3)δppm3.92(d,J=6.6Hz,2H),2.53-2.70(m,2H),2.32-2.52(m,2H),2.04(dp,J=13.3,6.7Hz,1H),1.64(t,J=1.5Hz,3H),1.01(d,J=6.7Hz,6H);MS(ES+)m/z169(MH+)。步骤2:在0℃向3-异丁氧基-2-甲基环戊-2-烯酮(34.5g)在DCM(300mL)中的溶液中滴加DIBAL-H(1M在己烷中,250mL)。将反应混合物在该温度搅拌90分钟。该反应用水淬灭,然后在DCM(200mL)和HCl溶液(1M,100mL)之间分配。水层用DCM萃取(2×200mL)。合并的有机层用饱和碳酸氢钠溶液(100mL)、盐水(200mL)洗涤,用Na2SO4干燥,过滤且真空浓缩以得到2-甲基环戊-2-烯醇和2-甲基环戊-2-烯酮(27.0g)的混合物,其为黄色油状物。步骤3:将2-甲基环戊-2-烯醇(27.0g)和氧化锰(IV)(5.0g)在乙醚(200mL)中的混合物在室温搅拌过夜。将混合物过滤且滤液真空浓缩。残余物真空蒸馏以得到2-甲基环戊-2-烯酮(16.5g),其为无色油状物。步骤4:向(R)-1-甲基-3,3-二苯基六氢吡咯并[1,2-c][1,3,2]氧杂氮杂硼杂环戊烯(oxazaborole)(1M在甲苯中,31.8mL)在无水THF(20mL)中的溶液中小心添加2-甲基环戊-2-烯酮(15.3g)和BH3(1M在THF中,111mL)。将混合物搅拌1小时。添加甲醇(150ml)且所得混合物用盐水稀释。水层用DCM萃取(2×200mL)。合并的有机层用Na2SO4干燥,过滤且真空浓缩以得到(S)-2-甲基环戊-2-烯醇(ee%未知,16.0g),其为黄色油状物。步骤5:在N2下向(S)-2-甲基环戊-2-烯醇(16.0g)和异二氢吲哚-1,3-二酮(36.0g)在THF(240mL)中的溶液中添加三苯基膦(77.0g)。将混合物冷却至0℃。将二异丙基偶氮二甲酸酯(63.4mL)滴加至混合物。搅拌30分钟后,将混合物在0℃搅拌过夜。去除溶剂且残余物通过柱色谱法纯化(用PE:EA=10:1洗脱)以得到粗产物,其通过SFC纯化以得到(R)-2-(2-甲基环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(10.6g,>98%ee),其为白色固体。手性HPLC(柱:AD-H,4.6*250mm,5μm,MeOH/CO2=10%,柱温度:40℃,CO2流速:2.7mL/min):tR=2.17分钟,ee%:>98%;1H-NMR(400MHz,CDCl3)δppm7.83(dd,J=5.4,3.1Hz,2H),7.76-7.61(m,2H),5.68(s,1H),5.21(d,J=7.4Hz,1H),2.69(dd,J=5.7,3.5Hz,1H),2.42-2.30(m,2H),2.22-2.12(m,1H),1.61(s,3H);MS(ES+)m/z228(MH+)。步骤6:向(R)-2-(2-甲基环戊-2-烯-1-基)异二氢吲哚-1,3-二酮(10.7g)在乙醇(150mL)中的溶液中添加肼.H2O(3.0mL)。回流3小时后,将反应混合物冷却至室温。将沉淀过滤且滤饼用EtOH(10mL)冲洗。向该滤液添加二噁烷中的HCl(4M,5mL)且将混合物浓缩。所得残余物溶于水,然后冷冻干燥以得到(R)-2-甲基环戊-2-烯胺的盐酸盐(6.3g),其为棕色固体,将其用于下一步而不用纯化。步骤7:向(R)-2-甲基环戊-2-烯胺盐酸盐(420mg)在甲苯(30mL)中的溶液中添加三光气(560mg)。所得混合物在110℃搅拌6小时。该混合物然后冷却至室温以得到(R)-5-异氰酸酯基-1-甲基环戊-1-烯的甲苯溶液。该溶液应每次新鲜合成。步骤8:在0℃向(R)-2-甲基环戊-2-烯胺盐酸盐(23mg)在DCM(3mL)和饱和NaHCO3水溶液(3mL)中的溶液中添加碳酸二(三氯甲基)酯(18mg)。在0℃至25℃将混合物搅拌2小时。分离所得两层,且水层用DCM萃取(60mL)。合并的有机层用盐水(15mL)洗涤,用Na2SO4干燥,过滤且在减压下浓缩以得到(R)-5-异氰酸酯基-1-甲基环戊-1-烯(20mg),其为白色固体,其直接用于下一步而不用进一步纯化。MS(ES+)m/z141(MH+)(M:含氢氧化铵的脲衍生物)。中间体F3:2-(叔丁基)-7-(叔丁基磺酰基)-6-氯苯并[d]噁唑步骤1:向2-(叔丁基)-6-氯苯并[d]噁唑-7-硫醇(3.0g)在DMF(30mL)中的溶液中添加2-碘丙烷(2.1g)。将混合物在100℃搅拌2小时。冷却后,去除溶剂。残余物通过柱色谱法纯化以得到2-(叔丁基)-6-氯-7-(异丙基硫基)苯并[d]噁唑(3.5g)。步骤2:在15℃向2-(叔丁基)-6-氯-7-(异丙基硫基)苯并[d]噁唑(3.5g)在DCM(40mL)中的溶液中添加mCPBA(5.3g)。将混合物在15℃搅拌48小时,然后用饱和Na2SO3溶液淬灭。有机层用Na2SO4干燥,过滤且浓缩。残余物通过柱色谱法纯化以得到2-(叔丁基)-6-氯-7-(异丙基磺酰基)苯并[d]噁唑(3.6g)。MS(ES+)m/z316(MH+)。步骤3:向2-(叔丁基)-6-氯-7-(异丙基磺酰基)苯并[d]噁唑(3.0g)在THF(10mL)中的溶液中添加LiHMDS(1M在THF中,31.7mL)。将混合物在-78℃搅拌10分钟,然后添加碘甲烷(6.74g)。将混合物在-78℃搅拌10分钟,然后用NH4Cl水溶液和HCl水溶液(10%)淬灭。将混合物用EA萃取。有机层用盐水洗涤,用Na2SO4干燥且浓缩。粗产物通过柱色谱法纯化以得到2-(叔丁基)-7-(叔丁基磺酰基)-6-氯苯并[d]噁唑(F3,2.8g)。MS(ES+)m/z330(MH+)。式(G)的化合物:1-(4-氯-2-羟基-3-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲三氟乙酸盐向6-氨基-3-氯-2-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯酚(中间体G1,80mg)在吡啶(5mL)中的溶液中滴加(R)-5-异氰酸酯基-1-甲基环戊-1-烯(中间体F2,74mg)在甲苯(5mL)中的溶液。将混合物在室温搅拌过夜。将混合物浓缩且所得残余物重溶于DMF(8mL)且通过MDAP纯化以得到1-(4-氯-2-羟基-3-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲的三氟乙酸盐(21mg),其为白色固体。1H-NMR(400MHz,DMSO-d6)δppm10.44(br.s.,2H),8.28(br.s.,1H),8.18(d,J=8.8Hz,1H),7.13(d,J=8.8Hz,1H),7.10(d,J=8.6Hz,1H),5.52(s,1H),4.49-4.62(m,2H),4.02(quin,J=7.5Hz,1H),2.82-3.23(m,2H),2.64-2.80(m,5H),2.11-2.36(m,3H),1.80-2.08(m,4H),1.67(s,3H),1.45-1.64(m,1H);MS(ES+)m/z454(MH+)。中间体G1:6-氨基-3-氯-2-(((1r,3r)-3-(吡咯烷-1-基)环丁基)磺酰基)苯酚步骤1:在0℃在氮气氛向顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁醇(中间体G2,3.0g)在DCM(30mL)中的溶液中添加TEA(2.6g)和甲烷磺酰氯(1.2g)。所得混合物在该温度搅拌30分钟。将混合物用NaHCO3水溶液淬灭,用DCM萃取(2×30mL)。洗涤合并的有机相,干燥且浓缩以得到甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯(2.6g)。MS(ES+)m/z422(MH+)。步骤2:向甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯(2.4g)在DMF(30mL)中的溶液中添加碳酸钾(1.6g)和吡咯烷(0.5g)。将混合物在80℃搅拌过夜。添加水(20mL)。粗产物用EA萃取(3×80mL)。合并的有机相用盐水洗涤,过滤且浓缩以得到粗产物,其通过柱色谱法纯化(用PE:EA=2:1洗脱)为2-(叔丁基)-6-氯-7-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯并[d]噁唑(2.0g)。MS(ES+)m/z397(MH+)。步骤3:将2-(叔丁基)-6-氯-7-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯并[d]噁唑(2.0g)溶于二噁烷的HCl中(50mL)和水(50mL)中。在120℃将反应混合物搅拌过夜。去除溶剂以得到粗产物(1.5g),将其与相同反应的另一批次合并,该反应使用2-(叔丁基)-6-氯-7-((反-3-(吡咯烷-1-基)环丁基)磺酰基)苯并[d]噁唑(1.3g)作为起始原料。混合物通过制备型HPLC纯化以得到标题化合物(614mg)。MS(ES+)m/z331(MH+)。中间体G2:顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁醇步骤1:向2-(叔丁基)-6-氯苯并[d]噁唑-7-硫醇(25.0g)在DMF(250mL)中的溶液中添加4-溴丁-1-烯(16.8g),然后添加K2CO3(21.4g)。所得混合物在60℃搅拌4小时。冷却后,将其倒入水(1L)中,且用EA萃取(2×200mL)。合并的有机层用水和盐水洗涤。用Na2SO4干燥后,将有机层浓缩以得到7-(丁-3-烯-1-基硫基)-2-(叔丁基)-6-氯苯并[d]噁唑(27.6g)。MS(ES+)m/z296(MH+)。步骤2:向冰-水冷却的7-(丁-3-烯-1-基硫基)-2-(叔丁基)-6-氯苯并[d]噁唑(27.6g)在DCM(400mL)中的溶液中分批添加mCPBA(84.0g)。在室温搅拌过夜后,添加NaHCO3水溶液和Na2S2O3水溶液。将混合物用DCM萃取(2×500mL)。合并的有机层用水和盐水洗涤,用Na2SO4干燥,过滤且浓缩。粗产物通过柱色谱法纯化(用PE:EA=1:0至7:3洗脱)以得到2-(叔丁基)-6-氯-7-((2-(氧杂环丙烷-2-基)乙基)磺酰基)苯并[d]噁唑(26.0g)。步骤3:在-70℃向2-(叔丁基)-6-氯-7-((2-(氧杂环丙烷-2-基)乙基)磺酰基)苯并[d]噁唑(10.0g)在THF(200mL)中的溶液中添加甲基溴化镁(3M在醚中,38.8mL)。将混合物缓慢加热且在室温搅拌过夜。将反应混合物倒入NH4Cl水溶液,且用EA萃取(2×200mL)。合并的有机层用盐水洗涤,用Na2SO4干燥且过滤。去除溶剂后,粗产物通过柱色谱法纯化(用PE:EA=4:1至3:2洗脱)以得到顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁醇(7.2g)。式(H)的化合物:1-(4-氯-3-((反-3-(二甲基氨基)环丁基)磺酰基)-2-羟基苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲三氟乙酸盐向6-氨基-3-氯-2-((反-3-(二甲基氨基)环丁基)磺酰基)苯酚(中间体H1,100mg)在吡啶(5mL)中的溶液中添加新鲜的在甲苯(5mL)中的(R)-5-异氰酸酯基-1-甲基环戊-1-烯(中间体F2,40mg)。将反应混合物在室温搅拌过夜,然后用水(5mL)淬灭。去除溶剂。残余物通过MDAP纯化(酸性条件)以得到1-(4-氯-3-((反-3-(二甲基氨基)环丁基)磺酰基)-2-羟基苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲的三氟乙酸盐(40mg)。1H-NMR(400MHz,DMSO-d6)δppm10.46(br.s.,1H),8.32(s,1H),8.16(d,J=8.8Hz,1H),7.06-7.17(m,2H),5.52(s,1H),4.50-4.58(m,1H),4.40-4.50(m,1H),3.98(quin,J=7.9Hz,1H),2.62-2.82(m,10H),2.11-2.36(m,3H),1.67(s,3H),1.49-1.59(m,1H);MS(ES+)m/z428(MH+)。中间体H1:6-氨基-3-氯-2-((反-3-(二甲基氨基)环丁基)磺酰基)苯酚步骤1:在室温将碳酸钾(262mg)添加至甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯(中间体H2;200mg)和二甲基胺盐酸盐(77mg)在DMF(3mL)中的溶液中。将反应混合物在100℃搅拌12小时,然后与相同反应的另一批次合并,该反应使用甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯(200mg)作为起始原料。合并的混合物用EA(50mL)稀释。有机相用水(2×20mL)和盐水(20mL)洗涤,用Na2SO4干燥且浓缩以得到反-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)-N,N-二甲基环丁胺(250mg),其为棕色油状物。MS(ES+)m/z371(MH+)。步骤2:在室温将HCl水溶液(35%,5mL)添加至反-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)-N,N-二甲基环丁胺(550mg)在1,4-二噁烷(10mL)和水(10mL)中的溶液中。将反应混合物在120℃搅拌3小时,然后与相同反应的另一批次合并,该反应使用反-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)-N,N-二甲基环丁胺(400mg)作为起始原料。将合并的混合物浓缩。残余物溶于MeOH。添加NaHCO3水溶液直到pH=8。将混合物浓缩。所得残余物通过柱色谱法纯化(用洗脱DCM:MeOH=50:1)以得到标题化合物(220mg),其为黑色油状物。MS(ES+)m/z305(MH+)。中间体H2:甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯在0℃向顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁醇(中间体G2,1.0g)和N,N-二乙基丙-2-胺(0.9mL)在DCM(20mL)中的溶液中添加甲磺酰氯(0.3mL)。将混合物在0℃搅拌4小时。然后将反应混合物倒入水中且用DCM萃取(2×50mL)。洗涤合并的有机相,干燥且浓缩以得到甲磺酸顺-3-((2-(叔丁基)-6-氯苯并[d]噁唑-7-基)磺酰基)环丁基酯(1.3g),其为无色固体。MS(ES+)m/z422(MH+)。式(I)的化合物:(R)-1-(4-氯-3-((1,1-二氟乙基)磺酰基)-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲向6-氨基-3-氯-2-((1,1-二氟乙基)磺酰基)苯酚(中间体I1,84mg)在吡啶(5mL)中的溶液中添加(R)-5-异氰酸酯基-1-甲基环戊-1-烯(中间体F2,0.15M在甲苯中,4mL)。将反应混合物在室温搅拌过夜。将混合物浓缩且残余物溶于DMF(8mL)且通过MDAP纯化以得到(R)-1-(4-氯-3-((1,1-二氟乙基)磺酰基)-2-羟基苯基)-3-(2-甲基环戊-2-烯-1-基)脲(15mg),其为白色固体。1H-NMR(400MHz,DMSO-d6)δppm10.53(br.s.,1H),8.28(s,1H),8.26(d,J=9.0Hz,1H),7.17(d,J=8.8Hz,1H),7.07(d,J=8.3Hz,1H),5.52(br.s.,1H),4.44-4.62(m,1H),2.23-2.36(m,2H),2.16-2.23(m,1H),2.09(t,J=19.2Hz,3H),1.67(s,3H),1.47-1.59(m,1H);MS(ES+)m/z395(MH+)。中间体I1:6-氨基-3-氯-2-((1,1-二氟乙基)磺酰基)苯酚步骤1:向在-78℃搅拌的2-(叔丁基)-6-氯苯并[d]噁唑-7-硫醇(6.0g)和氢氧化钾(13.9g)在乙腈(80mL)和水(80mL)中的溶液中一次性添加(溴代二氟甲基)膦酸二乙基酯(11.9g)。将混合物温热至室温且搅拌30分钟。添加EA(200mL)。分离有机相。水相用EA萃取(2×100mL)。合并有机层,用硫酸钠干燥,过滤且浓缩以得到2-(叔丁基)-6-氯-7-((二氟甲基)硫基)苯并[d]噁唑(7.2g),其为无色油状物。MS(ES+)m/z292(MH+)。步骤2:在0℃向2-(叔丁基)-6-氯-7-((二氟甲基)硫基)苯并[d]噁唑(7.2g)在DCM(200mL)中的溶液中添加mCPBA(19.5g)且搅拌2小时。由于起始原料没有消耗完全,添加另外的mCPBA(19.5g)。将混合物在室温搅拌过夜。然后添加Na2SO3水溶液。分离有机相,用饱和碳酸钠溶液和盐水洗涤。所得溶液用硫酸钠干燥,过滤且浓缩。残余物通过柱色谱法纯化(用PE:EA=1:0-2:3洗脱)以得到2-(叔丁基)-6-氯-7-((二氟甲基)磺酰基)苯并[d]噁唑(3.2g),其为白色固体。MS(ES+)m/z324(MH+)。步骤3:向2-(叔丁基)-6-氯-7-((二氟甲基)磺酰基)苯并[d]噁唑(2.2g)和碘甲烷(4.2mL)在THF(30mL)和HMPA(27mL)中的溶液中添加LDA(2M在THF中,13.5mL)。将混合物在-50℃搅拌30分钟。混合物然后用饱和NH4Cl溶液和10%HCl溶液中和。添加EA(50mL)。有机层用盐水洗涤,用硫酸钠干燥且真空浓缩。粗产物与相同反应的另一批次合并,该反应使用2-(叔丁基)-6-氯-7-((二氟甲基)磺酰基)苯并[d]噁唑(1.0g)作为起始原料。粗产物通过柱色谱法纯化(用PE:EA=1:0-3:2洗脱)以得到2-(叔丁基)-6-氯-7-((1,1-二氟乙基)磺酰基)苯并[d]噁唑(1.0g),其为白色固体。MS(ES+)m/z338(MH+)。步骤4:向2-(叔丁基)-6-氯-7-((1,1-二氟乙基)磺酰基)苯并[d]噁唑(1.0g)在1,4-二噁烷(20mL)中的溶液中添加浓HCl溶液(20mL)。混合物在110℃回流4小时,然后浓缩。所得残余物溶于EA(20mL)。溶液的pH用TEA调节至8。将混合物浓缩。残余物通过柱色谱法纯化(用PE:EA=1:0至1:4洗脱)以得到标题化合物(1.0g),其为浅棕色固体。MS(ES+)m/z272(MH+)。式(J)的化合物:1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲和式(C)的化合物:1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲向6-氨基-3-氯-2-((3-甲基四氢呋喃-3-基)磺酰基)苯酚(中间体JC1,3000mg)在吡啶(20mL)中的溶液中添加(R)-5-异氰酸酯基-1-甲基环戊-1-烯(中间体F2,2279mg)。所得混合物在40℃搅拌12小时。添加冷却水(30mL)且水层用DCM萃取(2×100mL)。合并的有机层用Na2SO4干燥,过滤且浓缩以得到1-(4-氯-2-羟基-3-((3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲(4.1g),其为黑色固体。一部分该化合物(3.0g)通过SFC和MDAP在酸性条件纯化以得到纯的两种对映异构体:1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲(式(J)的化合物,666mg)和1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-甲基环戊-2-烯-1-基)脲(式(C)的化合物,626mg)。式(J)的化合物:HPLC(ChiralpakIC柱(4.6*250mm,5uM),1:1ACN/IPA(包含0.5%DEA),CO2流速:2.55mL/min;共溶剂流速:0.45mL/min;反压:120巴);tr=16.9min;>93%ee;1H-NMR(400MHz,DMSO-d6)δppm10.53(s,1H),8.36(d,J=8.8Hz,1H),8.17(s,1H),7.14(d,J=8.8Hz,1H),7.05(d,J=8.3Hz,1H),5.51(s,1H),4.49-4.63(m,1H),4.33(d,J=10.3Hz,1H),3.76-3.89(m,2H),3.61(d,J=10.0Hz,1H),2.68(dt,J=13.3,7.9Hz,1H),2.10-2.35(m,3H),1.91-2.02(m,1H),1.67(s,3H),1.44-1.58(m,4H);MS(ES+)m/z415(MH+);式(C)的化合物:HPLC(ChiralpakIC柱(4.6*250mm,5uM),1:1ACN/IPA(包含0.5%DEA),CO2流速:2.55mL/min;共溶剂流速:0.45mL/min;反压:120巴);tr=14.3min;>99%ee;1H-NMR(400MHz,DMSO-d6)δppm10.52(s,1H),8.36(d,J=9.0Hz,1H),8.17(s,1H),7.14(d,J=8.8Hz,1H),7.05(d,J=8.3Hz,1H),5.51(s,1H),4.49-4.61(m,1H),4.33(d,J=10.0Hz,1H),3.75-3.91(m,2H),3.61(d,J=10.0Hz,1H),2.68(dt,J=13.3,7.9Hz,1H),2.09-2.37(m,3H),1.90-2.02(m,1H),1.67(s,3H),1.42-1.59(m,4H);MS(ES+)m/z415(MH+);中间体JC1:6-氨基-3-氯-2-((3-甲基四氢呋喃-3-基)磺酰基)苯酚步骤1:向四氢呋喃-3-醇(5.0g)在DCM(100mL)中的冰-水冷却的溶液中添加TEA(11.9mL)和MsCl(4.9mL)。所得反应混合物在0℃搅拌3小时,然后用NaHCO3水溶液淬灭。将混合物用EA萃取(2×100mL)。将合并的有机相洗涤,干燥,过滤且浓缩以得到甲磺酸四氢呋喃-3-基酯(6.2g)。步骤2:向2-(叔丁基)-6-氯苯并[d]噁唑-7硫醇钠(11.8g)在DMF(100mL)中的溶液中添加甲磺酸四氢呋喃-3-基酯(6.2g)和K2CO3(7.7g)。所得反应混合物在80℃搅拌过夜。冷却后,将其倒入水(500mL)中且用EA萃取(2×150mL)。将合并的有机相洗涤,干燥且浓缩以得到2-(叔丁基)-6-氯-7-((四氢呋喃-3-基)硫基)苯并[d]噁唑(11.4g)。步骤3:向2-(叔丁基)-6-氯-7-((四氢呋喃-3-基)硫基)苯并[d]噁唑(7.4g)在DCM(200mL)中的冰-水冷却的溶液中添加mCPBA(11.7g)。所得混合物在室温搅拌2天,然后用NaHCO3水溶液和Na2S2O3水溶液淬灭。将混合物用EA萃取(2×200mL)。将合并的有机相洗涤,干燥且浓缩。残余物通过柱色谱法纯化(用PE中的0-40%EA洗脱)以得到2-(叔丁基)-6-氯-7-((四氢呋喃-3-基)磺酰基)苯并[d]噁唑(4.3g)。MS(ES+)m/z344(MH+)。步骤4:向2-(叔丁基)-6-氯-7-((四氢呋喃-3-基)磺酰基)苯并[d]噁唑(4.3g)和MeI(2.0mL)在THF(100mL)中的干冰-乙醇冷却的溶液中添加LiHMDS(1M在THF中,31.3mL)。所得混合物缓慢加热且在室温搅拌30分钟。添加NH4Cl水溶液。将混合物用EA萃取(2×150mL)。将合并的有机相洗涤,干燥且浓缩以得到2-(叔丁基)-6-氯-7-((3-甲基四氢呋喃-3-基)磺酰基)苯并[d]噁唑(4.4g)。MS(ES+)m/z358(MH+)。步骤5:向2-(叔丁基)-6-氯-7-((3-甲基四氢呋喃-3-基)磺酰基)苯并[d]噁唑(4.4g)在1,4-二噁烷(150mL)中的溶液中添加HCl水溶液(37%,30.3mL)。将混合物在120℃回流过夜,然后浓缩。残余物溶于水(100mL)。溶液的pH用NaHCO3水溶液调节至8,且用EA萃取。将有机相洗涤且浓缩。所得残余物通过柱色谱法纯化(用PE中的0-80%EA的梯度洗脱)以得到标题化合物(2.4g)。MS(ES+)m/z292(MH+)。式(K)的化合物:1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲和式(L)的化合物:1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲向6-氨基-3-氯-2-((3-甲基四氢呋喃-3-基)磺酰基)苯酚(中间体JC1,600mg)在吡啶(20mL)中的溶液中添加在甲苯(20mL)中的新鲜的(R)-1-氯-5-异氰酸酯基环戊-1-烯(中间体E2,443mg)。将混合物在室温搅拌过夜。所得溶液用水(5mL)淬灭且浓缩。残余物用MDAP纯化(酸性条件)以得到1-(4-氯-2-羟基-3-((3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲(205mg)。将一部分该化合物(160mg)通过手性分离纯化(AD-H(4.6*250mm,5um),共溶剂MeOH,1%DEA),然后通过MDAP(酸性条件)纯化以得到1-(4-氯-2-羟基-3-(((S)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲和1-(4-氯-2-羟基-3-(((R)-3-甲基四氢呋喃-3-基)磺酰基)苯基)-3-((R)-2-氯环戊-2-烯-1-基)脲(分别为式(K)的化合物和式(L)的化合物,37mg和31mg),其为紫罗兰色固体。异构体1:HPLC(AD-H柱(4.6*250mm,5uM),IPA(包含0.1%DEA),CO2流速:2.25mL/min;共溶剂流速:0.75mL/min;反压:149巴);tr=4.7min;>99%ee;1H-NMR(400MHz,DMSO-d6)δppm10.49(s,1H),8.37(d,J=8.9Hz,1H),8.25(s,1H),7.38(d,J=8.8Hz,1H),7.16(d,J=8.9Hz,1H),5.99(d,J=1.7Hz,1H),4.73(s,1H),4.34(d,J=10.1Hz,1H),3.72-3.97(m,2H),3.61(d,J=10.1Hz,1H),2.67(m,1H),2.18-2.47(m,3H),1.84-2.04(m,1H),1.57-1.78(m,1H),1.49(s,3H);MS(ES+)m/z435(MH+)。异构体2:HPLC(AD-H柱(4.6*250mm,5uM),IPA(包含0.1%DEA),CO2流速:2.25mL/min;共溶剂流速:0.75mL/min;反压:149巴);tr=5.5min;>93%ee;1H-NMR(400MHz,DMSO-d6)δppm10.49(s,1H),8.37(d,J=8.9Hz,1H),8.24(s,1H),7.37(d,J=8.7Hz,1H),7.16(d,J=8.9Hz,1H),5.99(s,1H),4.73(s,1H),4.34(d,J=10.1Hz,1H),3.71-3.96(m,2H),3.61(d,J=10.1Hz,1H),2.62-2.78(m,1H),2.19-2.44(m,3H),1.84-2.06(m,1H),1.56-1.78(m,1H),1.49(s,3H);MS(ES+)m/z435(MH+)。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1