一种质粒载体及其构建方法和应用与流程
本发明属于微生物技术领域,具体涉及一种质粒载体及其构建方法和应用。
背景技术:
葡萄座腔菌(botryosphaeriadothidea)是一种常见的全球分布的重要真菌,具有重要的开发利用价值。这类真菌有的可以腐生在各种腐木上,有的在多种木本植物上寄生,引起木本植物的病害,有的与植物形成共生体,是许多植物的内生真菌。在腐木上营腐生生活的葡萄座腔菌可以将木本植物残体分解,在生态系统的物质循环和能量循环中具有重要作用;同时,这类真菌还可以被开发以生产木质素酶和纤维素酶,具有很好的经济开发前景。而属于内生真菌的菌株可以作为生物反应器,如从北非雪松中分离的一株真菌可以产生15种新的化合物,其中有3种具有抗病原真菌、抗氧化功能。
作为植物病原菌的葡萄座腔菌菌株也有开发价值,是重要的待开发生物资源。植物病原菌要成功侵染寄主植物,就必须能够克服植物的防卫反应,还要能够穿过植物表皮等植物的物理保护屏障。因此,植物病原菌往往能够产生比腐生菌更多的胞外降解酶和其他效应子,使植物的防卫反应下降,并将植物表皮细胞壁成分进行必要的降解。
一些真菌的基因组序列分析结果也表明,与腐生真菌相比,植物病原真菌的基因组中含有更多的细胞壁降解酶基因,而内生真菌的基因组中含有的细胞壁降解酶基因最少。因此,植物病原菌可以开发成生产胞壁降解酶类的生物资源,如降解作物秸秆或林木残体,在环境保护、生物质能源开发方面具有很好的应用前景。作为植物病原菌的葡萄座腔菌菌株还可以用于生物转化生产特定的功能化合物,如诺卡酮(nootkatone)是重要的工业原料,葡萄座腔菌可以将巴伦西亚橘烯转化为诺卡酮nootkatone,在减肥产品、增强免疫力的保健产品和美容产品。
随着现代生物技术的发展,分子生物学技术已经与传统生物学技术结合在一起,大大促进了生物学研究和生物产业的发展,无论是对生物细胞的分子改造,还是克隆有用的基因,都需要建立外源基因转化进入细胞的转化体系,这是对细胞进行分子改造和研究的前提。针对葡萄座腔菌这种具有重要经济意义的真菌,截止目前,该菌的基因转化效率比较低。
例如,质粒pko1-hph是丝状真菌基因转化中常用的载体,可以用农杆菌介导的方法,对多种子囊菌进行基因转化。其上含有一个由trpc启动子控制的抗潮霉素基因作为选择标记,还带有一个由稻梨形孢菌组蛋白h3基因启动子控制的绿色荧光蛋白(gfp)基因作为报告基因。但是本课题组在前期研究中用这个质粒转化葡萄座腔菌时,发现转化效率低,绿色荧光蛋白表达很弱。
为此,本课题组构建了新的筛选标记,并整合到可用于农杆菌转化的载体上,建立了利用农杆菌介导的外源dna高效转化体系,为该类真菌的遗传改造、开发有用的基因、分离病原真菌的药物靶点基因、研究该菌的基因功能等方面的研究开发提供了可行性方案。
技术实现要素:
本发明提供了一种质粒载体,解决了现有质粒转化到葡萄座腔菌时,转化效率低的问题。
一种质粒载体,由pcambia1300改造构建形成,在pcambia1300多克隆位点插入葡萄座腔菌组蛋白h3基因启动子、葡萄座腔菌转录延长因子α基因启动子、抗性基因,所述抗性基因位于葡萄座腔菌转录延长因子α基因启动子的下游。
质粒pcambia1300最早用于多种植物的农杆菌介导的遗传转化,后来用于农杆菌介导的遗传转化的多种质粒载体也是在这个载体的基础上构建而成,本发明也是采用pcambia1300作为原始载体。
一般真核生物细胞中,组蛋白h3基因和转录延长因子α基因在细胞分裂和rna生物合成中具有重要功能,这两个基因的启动子在细胞中都是强启动子。本发明选择葡萄座腔菌组蛋白h3基因启动子作为调控外源基因(如gfp)高表达的强启动子,而把葡萄座腔菌转录延长因子α基因启动子作为转化葡萄座腔菌细胞时筛选标记的强启动子。
本发明研究证明,构建的质粒载体转化到葡萄座腔菌中的转化效率远高于目前常用的转化载体。以目前常用的载体pko1-hph为例,在相同的转化条件下,以pko1-hph作为载体获得的转化子平均只有3个时,利用本发明的质粒载体能够获得13~20个转化子。
作为优选,所述抗性基因为潮霉素抗性基因。原始载体pcambia1300中即含有潮霉素抗性基因,但是其启动子为35s启动子,在真菌中表达效率很低,在葡萄座腔菌细胞中几乎不表达。研究发现,与对照载体pko1-hph比较,本发明构建的质粒载体中潮霉素抗性基因的表达量高出10倍以上,证明葡萄座腔菌转录延长因子α基因启动子调控潮霉素抗性基因在葡萄座腔菌细胞中高表达,有利于转化子的筛选。
作为优选,在葡萄座腔菌组蛋白h3基因启动子的下游插入报告基因,可以通过报告基因来标定目的基因的表达,所述目的基因可以是gfp基因。
本发明具体提供两个质粒,具体为:
一种质粒载体,命名为pbdh3g-thy1,大小为11219bp,其中葡萄座腔菌组蛋白h3基因启动子的核苷酸序列如seqidno.1所示,所述葡萄座腔菌转录延长因子α基因启动子的核苷酸序列如seqidno.2所示。
另一种质粒载体,命名为pbdh3g-thy2,大小为10217bp,与pbdh3g-thy1的区别在于,只用了葡萄座腔菌转录延长因子α基因启动子dna片段的一部分。所述葡萄座腔菌转录延长因子α基因启动子的核苷酸序列如seqidno.3所示。
本发明还提供了构建所述质粒载体的构建方法,包括以下步骤:
(1)扩增分别包含所述葡萄座腔菌组蛋白h3基因启动子、所述葡萄座腔菌转录延长因子α基因启动子和所述抗性基因的dna片段;
(2)通过重组反应将所述dna片段插入到pcambia1300载体,得到所述的质粒载体。
具体是通过设计特异性引物使得扩增出的pcr产物的5’和3’最末端分别带有与相邻片段最末端对应的完全一致的序列,在重组酶的作用下,完成定向克隆。
作为优选,所述葡萄座腔菌组蛋白h3基因启动子下游插入gfp基因,所述抗性基因为 潮霉素抗性基因,设计引物如下:
扩增葡萄座腔菌组蛋白h3基因启动子的引物为:
上游引物:cattattatggagaaacggcagcagaaagggttgagac;
下游引物:cctcgcccttgctcaccatgcagaagttgtgttgggtcgg;
扩增gfp基因的引物为:
上游引物:atggtgagcaagggcgaggag;
下游引物:tctagattacttgtacagctcgtccatgccg;
扩增葡萄座腔菌转录延长因子α基因启动子的引物为:
上游引物:ctgtacaagtaatctagatcgagagtggagagtggcgagaaa;
下游引物:gtgagttcaggctttttcattgtagaggctgaggtgtctg;
或
上游引物:ctgtacaagtaatctagaggagtcataccgtgaaca;
下游引物:gtgagttcaggctttttcattgtagaggctgaggtgtctg;
扩增抗性基因的引物为:
上游引物:atgaaaaagcctgaactcaccg;
下游引物:acgacggccagtgccacatctactctattcctttgccc。
本发明还提供了一种农杆菌介导的葡萄座腔菌基因转化方法,包括以下步骤:
(1)将目的基因插入所述质粒载体中,转化到农杆菌中,得到含有重组质粒的农杆菌;
(2)将含有重组质粒的农杆菌与恢复培养后的葡萄座腔菌原生质体共培养,获得转化子。
利用农杆菌介导技术,本发明构建的质粒载体能够稳定有效地将目的基因整合到葡萄座腔菌的基因组中,并且使得目的基因在葡萄座腔菌细胞中高效的表达。以表达外源基因gfp为例,本发明构建的质粒载体使gfp基因在转化子中的表达量高于对照pko1-hph的46倍。为葡萄座腔菌的基因功能研究、遗传改造、开发有用基因等方面研究提供技术支持。
本发明具备的有益效果:
(1)本发明通过在原始载体pcambia1300的多克隆位点插入依次连接的葡萄座腔菌组蛋白h3基因启动子、葡萄座腔菌转录延长因子α基因启动子、抗性基因,形成新的质粒载体;选择的葡萄座腔菌组蛋白h3基因和转录延长因子α基因启动子的区段,能够高效启动连接在后面的基因,因此大大提高该质粒载体的转化效率高。
(2)本发明建立了利用农杆菌介导的外源基因高效转化葡萄座腔菌的方法,为该菌的遗传改造、开发有用基因、分离病原真菌的药物靶点基因、研究该菌的基因功能等方面的研究开发提供可行性方案。
附图说明
图1为载体pbdh3g-thy1的结构示意图。
图2为三种载体的转化葡萄腔菌的转化率。
图3为三种载体的转化子的菌丝体中在荧光视野和白光视野下的状态,其中a为载体 pbdh3g-thy1(a1是荧光视野下的状态,a2是白光视野下的状态);b为载体pbdh3g-thy2(b1是荧光视野下的状态,b2是白光视野下的状态);c为载体pko1-hph(c1是荧光视野下的状态,c2是白光视野下的状态)。
图4为载体pbdh3g-thy1的转化子中,gfp在分生孢子、萌发、分生孢子器细胞中的表达情况,其中a为分生孢子(a1是荧光视野下的状态,a2是白光视野下的状态);b为正在萌发的分生孢子(b1是荧光视野下的状态,b2是白光视野下的状态);c为分生孢子器(c1是荧光视野下的状态,c2是白光视野下的状态)。
图5为随机转化子中抗潮霉素基因片段的pcr扩增结果,其中m为分子量标记;p为阳性对照;w为野生型菌株;1-12为转化子。
图6为随机转化子中gfp基因片段的pcr扩增结果,其中m为分子量标记;p为阳性对照;w为野生型菌株;1-12为转化子。
图7为三种载体的转化子中抗潮霉素基因的相对表达量。
图8为三种载体的转化子中gfp基因的相对表达量。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
实施例1用于高效转化葡萄座腔菌的质粒载体pbdh3g-thy的构建
本发明质粒pegfp购买自clontech公司;质粒pcambia1300信息:hajdukiewiczp,svabz,maligap(1994)thesmall,versatileppzpfamilyofagrobacteriumbinaryvectorsforplanttransformation.plantmolbiol25:989-994.;质粒pcb1003信息:carrollam,sweigardja,barbaravc(1994)improvedvectorsforselectingresistancetohygromycin.fungalgeneticnews,41:22.上述3个质粒都保存在浙江大学农学院真菌生物学实验室。
在葡萄座腔菌基因组数据库(http://genome.jgi.doe.gov/botdo1/botdo1.home.html)中进行检索,找到葡萄座腔菌的组蛋白h3基因和转录延长因子α(tef)基因,分别将其上游约1500bp(含有各自基因的启动子)下载,然后设计引物。再以质粒pcb1003上的潮霉素抗性基因序列,设计可以通过pcr方法扩增潮霉素抗性基因的引物,以质粒pegfp上的gfp基因序列,设计可以通过pcr方法gfp基因的引物。各个引物的序列见表1(由上至下依次如seqidno.4~22所示)。
表1所用的引物序列信息
(1)通过pcr扩增获得葡萄座腔菌组蛋白h3基因启动子(h3promoter):
pcr扩增反应在朗基mg96g型pcr仪上进行,其中,pcr反应体系(50μl):引物hp1和bd-h3prd各2μm,dntps200μm,mg2+1.5mm,5×pspcrbuffer10μl,基因组dna10ng,primestardna聚合酶2u。
pcr反应条件:94℃预变性2min,然后35个循环包括:94℃变性30sec,59℃退火40sec,72℃延伸1.5min。最后72℃后延伸10min。
(2)通过pcr扩增获得葡萄座腔菌转录延长因子α(tef)基因启动子(tefpromoter):
pcr扩增反应在朗基mg96g型pcr仪上进行,其中,pcr反应体系(50μl):引物tp1(tp4)和bdtefpdh各2μm,dntps200μm,mg2+1.5mm,5×pspcrbuffer10μl,基因组dna10ng,primestardna聚合酶2u。
pcr反应条件:94℃预变性2min,然后35个循环包括:94℃变性30sec,56℃退火40sec,72℃延伸1.5min。最后72℃后延伸10min。
(3)通过pcr扩增获得gfp基因(gfp):
pcr扩增反应在朗基mg96g型pcr仪上进行,其中,pcr反应体系(50μl):引物egfpu和egfpdxz各2μm,dntps200μm,mg2+1.5mm,5×pspcrbuffer10μl,模板(质粒pegfp)dna1ng,primestardna聚合酶2u。
pcr反应条件:94℃预变性2min,然后35个循环包括:94℃变性30sec,55℃退火40sec,72℃延伸1min。最后72℃后延伸10min。
(4)通过pcr扩增获得抗潮霉素基因(hygromycinb):
pcr扩增反应在朗基mg96g型pcr仪上进行,其中,pcr反应体系(50μl):引物hyg2和hph3各2μm,dntps200μm,mg2+1.5mm,5×pspcrbuffer10μl,模板(质粒pcb1003)dna1ng,primestardna聚合酶2u。
pcr反应条件:94℃预变性2min,然后35个循环包括:94℃变性30sec,57℃退火40sec,72℃延伸1min。最后72℃后延伸10min。
(5)载体的构建:
用限制性内切酶xhoⅰ和hindⅲ酶切质粒pcambia1300,胶回收6.8kb的大片段。然后将上述获得的pcr片段用pcr片段纯化试剂盒纯化后,通过一步克隆方法(vazyme公司的cloneexpressmultis试剂盒),通过重组反应构建新的载体,重组体系为:5×cemultisbuffer4μl,pcambia1300酶切产物50ng,h3promoter、gfp、tefpromoter、hygromycinb各25ng,exnasemultis酶2μl,用ddh2o补足20μl,混匀后37℃保温30min。
重组好的载体用常规方法转化大肠杆菌dh5α:取20μl冷却的反应液,加入到200μl感受态细胞中,轻弹管壁数下混匀,在冰上放置30min。42℃热激45~90秒,冰水浴孵育2min。加入900μl的lb培养基,37℃孵育10min充分复苏。37℃摇菌45min。然后离心沉淀菌体,取100μllb培养液重新悬菌液后,全部均匀涂布在含有50μg/ml的卡那霉素的平板上。将平板倒置,于37℃过夜培养。
通过是菌落pcr进行克隆的鉴定。用无菌的枪头或牙签将单个菌落挑至20μl的lb培养基中混匀,直接取1μl作为pcr模板。分别用(3)(4)方法pcr扩增gfp和hygromycinb基因。将pcr阳性菌落的剩余菌液接种至含有50μg/ml的卡那霉素的lb培养基中培养过夜,取100lb送生工生物技术有限公司进行测序验证,然后在2ml菌液中加入甘油,使甘油浓度为25%,置于-80℃超低温冰箱中保存备用。
构建的质粒载体定名为pbdh3g-thy1,共11.2kb(图1),我们同时构建了pbdh3g-thy2载体,这个载体与pbdh3g-thy1区别是只用了tefpromoter的一部分,即在步骤(2)中,用tp4代替tp1进行pcr,其他构建过程相同,pbdh3g-thy2载体大小为10.9kb。
实施例2葡萄座腔菌原生质体制备
1、试剂和培养基的制备
(1)0.7m氯化钠溶液:40.95g氯化钠(nacl)溶解在1000ml水中,定容后后进行常规的高温灭菌(1.1个大气压,121℃下灭菌20min)。
(2)1mtris-cl,ph=7.5:三羟甲基氨基甲烷(tris(hydroxymethyl)aminomethane,一般简称为tris)121.14溶于600ml重蒸水中,用浓盐酸调整ph=7.5,然后用重蒸水定容至1l,进行常规的高温灭菌(1.1个大气压,121℃下灭菌20min),备用。
(3)细胞壁降解酶液:在天平上称取100mg的崩溃酶(driselase)和100mg的溶壁酶(lysingenzyme),溶解在10ml的0.7m氯化钠溶液中,用直径为0.22μm的滤膜过滤除菌,现配现用。
(4)100ml的stc溶液:称取21.8604g的山梨醇(sorbitol),0.735g的氯化钙加水约60ml 溶解后,加入配好的1mtris-cl(ph=7.5)1ml,然后定容至100ml。
(5)马铃薯葡萄糖琼脂培养基(potatodextroseagar,pda):在马铃薯200g切成小块,加水煮沸15min,然后2层纱布过滤后,弃去马铃薯块,在过滤液中加入葡萄糖20g、琼脂20g中加蒸馏水定容至1000ml;然后进行常规的高温灭菌(1.1个大气压,121℃下灭菌20min)。
(6)马铃薯葡萄糖液体培养基(potatodextrosebroth,pdb):在马铃薯200g切成小块,加水煮沸15min,然后2层纱布过滤后,弃去马铃薯块,在过滤液中加入葡萄糖20g,加蒸馏水定容至1000ml;然后进行常规的高温灭菌(1.1个大气压,121℃下灭菌20min)。
2、葡萄座腔菌原生质体的制备
(1)将本实验室保存的葡萄座腔菌菌株fy-31接种到pda平板上,25℃培养5天后,用挑针挑取5-8块约3×3mm2的菌丝块,接种到100ml的pdb液体培养基中,25℃培养2天。
(2)在超净工作台中,用四层纱布过滤菌丝,然后用0.7mnacl冲洗菌丝3次,用灭菌的滤纸和吸水纸滤干水分,称取1g菌丝置于50ml灭菌离心管中,加入10ml配好的细胞壁降解酶液,在28℃的摇床上以80转/min的转速酶解3.5小时。
(3)用灭菌的两层擦镜纸过滤酶解液,再用0.7mnacl冲洗残留在擦镜纸上的原生质体两次,每次用10ml。除去残渣,将过滤液转移到50ml离心管中,置于离心机中,在4℃条件下,用3000rpm离心10min。
(4)小心弃上清,加入10-20mlstc,轻轻晃动离心管以悬浮沉淀。
(5)将50ml离心管置于离心机中,在4℃条件下,用3000rpm离心10min。
(6)小心弃上清,加入1mlstc,轻轻晃动离心管以悬浮沉淀。
(7)用血球计数板对原生质体计数,根据计数结果,加入适量stc溶液,调整原生质体的浓度为106个/ml。
(8)将获得的原生质体分装到已经灭菌的1.5ml离心管中,每管100ul,-80℃保存备用。
实施例3农杆菌介导的葡萄座腔菌dna转化
1、葡萄座腔菌菌株fy-31的准备:
将本实验室保存的葡萄座腔菌菌株fy-31接种到pda平板上,25℃培养5天,备用。
2、质粒的准备
提取实施例1中的pbdh3g-thy1和pbdh3g-thy2,以及用于对照的pko1-hph质粒,将质粒的浓度调整到0.25μg/μl。
3、用冻融法将质粒载体转化到农杆菌中
(1)lb固体培养基配方如下:胰蛋白胨(trypone)10g,酵母提取物(yeastextract)5g,氯化钠(nacl)10g,琼脂15g,水1000ml,ph=7.0。常规的高温灭菌(1.1个大气压,121℃下灭菌20min)。
lb液体培养基:lb固体培养基中不加琼脂,其他相同。
(2)将农杆菌菌株agl1在lb平板上划线活化,28℃培养2天。然后将1个单菌落转到5mllb液体培养基中28℃振荡培养过夜。
(3)将2ml培养液转移到装有50ml的lb液体培养基的三角瓶中,28℃振荡培养6小时,测定其吸光度,当od600=0.5~1.0时停止培养。
(4)将培养好的菌液放置在冰上,然后转移到50ml离心管中,在4℃条件下用3000g离心5min。
(5)弃去上清液,沉淀用1ml预冷的20mmcacl2溶液悬浮,分装到预冷的1.5ml离心管中,每管0.1ml。
(6)取浓度调整到0.25μg/μl的三种质粒4μl分别加入到含有0.1ml农杆菌的1.5ml离心管中,盖好盖子,轻轻混匀,然后迅速放到液氮中冷冻。
(7)取出1.5ml离心管,置于37℃水浴中保温5min解冻。
(8)在1.5ml离心管中加入1ml的lb培养液,在28℃按100rpm轻摇培养2~4h。
(9)将离心管放入离心机中,12000rpm离心2min,弃去1ml培养液,然后将沉淀悬浮,涂布于含有卡那霉素的lb平板上,正面放置30min,待菌液完全被培养基吸收后,倒置培养皿,28℃培养2~4天。
4、利用农杆菌转化真菌葡萄座腔菌菌株fy-31
农杆菌培养基im配方:0.8ml1.25k-phosphate-bufferph4.8(用kh2po4和k2hpo4配制);20mlmn-buffer(30g/lmgso4·7h2o,15g/lnacl);1ml1%cacl2·2h2o(w/v);10ml0.01%feso4(w/v);5mlsporeelements(100mg/lznso4·7h2o,100mg/lcuso4·h2o,100mg/lh3bo3,100mg/lna2moo4·2h2o)(过滤灭菌);2.5ml20%nh4no3(w/v);10ml50%glycerol(v/v);40ml1mmesph5.5(用naoh调ph值);20%glucose(w/v),液体培养基中加10ml,固体培养基加5ml;加水至1l,固体培养基加1.5%琼脂粉。
100mm乙酰丁香酮储备液(acetosyringone,as):1.962g乙酰丁香酮用二甲基亚砜(dmso)溶解,定容到100ml,用孔径为0.22μm的微孔滤膜过滤除菌。
农杆菌诱导培养基aim配方:在100ml的im培养基中加入乙酰丁香酮储备液200μl,使乙酰丁香酮的终浓度为200μm。
恢复培养基yps(yeastextractpeptonesucrose):蛋白胨(peptone)5g,酵母提取物(yeastextract)3.5g,葡萄糖(glucose)10g,蔗糖(sucrose)342.3g,水1000ml,ph=7.0。常规的高温灭菌(1.1个大气压,121℃下灭菌20min)。
卡纳霉素储备液(50mg/ml):将1g氯霉素溶解在20ml的无水乙醇中备用。
链霉素储备液(10mg/ml):将100mg链霉素溶解在10ml的灭菌水中备用。
头孢霉素储备液(400mg/ml):将2g头孢霉素溶解在5ml的灭菌水中备用
潮霉素储备液:50mg/ml,由生工生物技术有限公司购买。
转化步骤:
(1)从新鲜培养的lb平板(含50μg/ml卡那霉素)上挑选一农杆菌单菌落接种于5mllb液体培养基(含50μg/ml卡那霉素)中,200rpm/min,28℃过夜培养。
(2)第二天,将400μl培养液转移到5ml含50g/ml卡那霉素的诱导液体培养基(aim)中,od值约0.15,28℃培养5~6个小时,使菌液的od600达到0.5~0.6。
(3)葡萄座腔菌菌株fy-31转化材料的优化:在含有100μl原生质体的1.5ml离心管中加入500μl的恢复培养基,28℃恢复培养5~6小时。
(4)将100ml的im固体培养基中融化后,在室温冷却到50℃,然后加入乙酰丁香酮储备液200μl,头孢霉素储备液100μl,链霉素储备液100μl,卡那霉素储备液100μl,轻轻摇动混匀后,倒入6cm一次性塑料培养皿中,制成诱导平板。待培养皿中的培养基凝固后,将一张直径5cm的灭菌纤维素膜覆盖在培养基表面。
(5)取100μl培养好的农杆菌agl-1(含载体)菌液和200μl转化材料混合,混合液均匀涂于诱导平板上的硝酸纤维素膜表面,22℃共培养48h。
(6)将100ml的pda固体培养基中融化后,在室温冷却到50℃,然后加入乙酰丁香酮储备液200μl,头孢霉素储备液100μl,链霉素储备液100μl,卡那霉素储备液100μl,潮霉素储备液20μl,轻轻摇动混匀后,倒入6cm一次性塑料培养皿中,制成筛选平板。将诱导平板上的硝酸纤维素膜切成约0.5cm的条带,转移到筛选平板上,条带之间相隔约0.5cm,置于在28℃培养7天,可以见到有菌丝长到硝酸纤维素膜条带之间的培养基上。
(7)挑取长出的菌丝,转移到含有10μg/ml潮霉素的pda平板上,28℃培养5天,确认是否转化成功。如果能够生长,可确认转化成功。
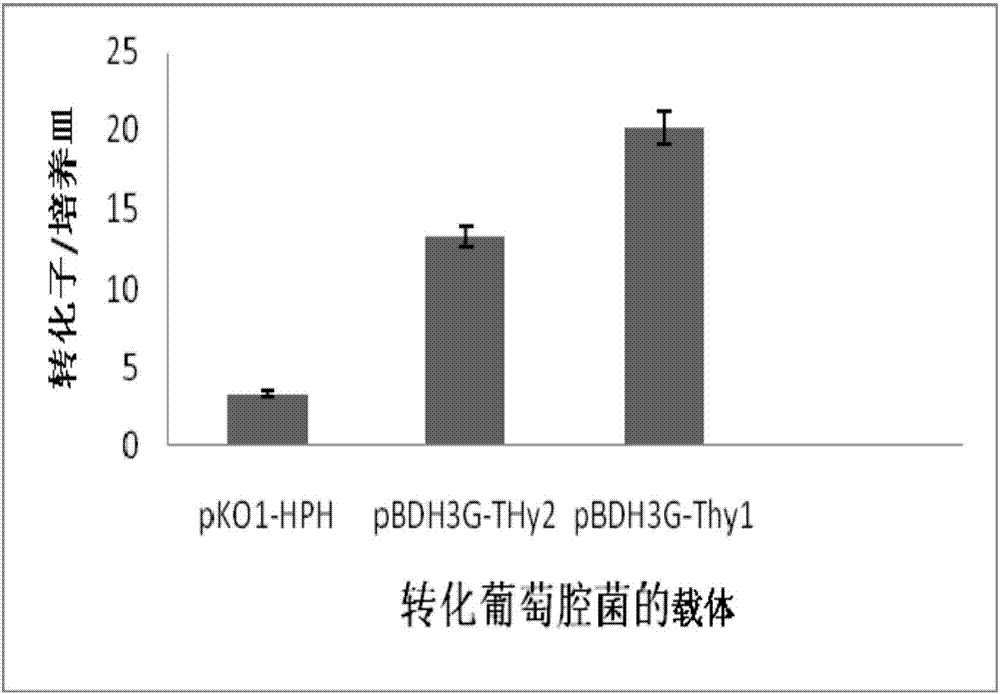
转化结果如图2所示:载体pbdh3g-thy1平均每个培养基可以获得20转化子以上pbdh3g-thy2可以获得13个转化子以上,而对照pko1-hph平均每个培养皿仅获得约3个转化子。结果表明本发明构建的载体转化效率比其它载体的转化效率大大提高。
实施例4转化子的检测
1、葡萄座腔菌转化子的稳定性检测
从获得的转化子中随机挑选了12个转化子,在pda培养基平板上28℃转接培养5次,然后再转接在含有10μg/ml潮霉素的pda平板上,28℃培养5天。
结果这12个转化子都可以生长,说明转化子的稳定性好,转入的抗性基因稳定存在。
2、葡萄座腔菌转化子的绿色荧光检测
随机选择三个载体转化的葡萄座腔菌转化子在pda平板上,28℃培养5天。用挑针挑取少量菌丝,置于载玻片上,制成观察玻片,放到显微镜的载物台上。打开紫外光(450~490nm),观察绿色荧光。
结果如图3所示,本发明构建的载体pbdh3g-thy1和pbdh3g-thy2的转化子的菌丝体也可以发出较强的荧光,而作为对照的pko1-hph的转化子只有少部分菌丝发出很弱的荧光。如图4所示,本发明构建的载体的转化子在分生孢子、孢子萌发、菌丝体、以及分生孢子器细胞中都能表达,说明本发明构建的载体可以使外源基因在葡萄座腔菌细胞中高效的表达。
3、葡萄座腔菌转化子的pcr检测。
(1)真菌基因组dna的小量提取
提取缓冲液:含有1mkcl、100mmtris-hcl、10mmedta,ph=8.0。
(2)提取方法:
①将随机挑选的12个转化子菌株在pda平板上25℃生长7天后,用牙签刮取平板上的菌丝,放入已灭菌的含有300μl提取缓冲液1.5ml离心管中;②用电磨机将挑取的菌丝磨碎,然后再加入300μl提取缓冲液,剧烈震荡2min;
③10000rpm离心10min;
④吸取上清至另一离心管中,弃沉淀;
⑤加入等体积的异丙醇(分析纯),沉淀核酸,轻轻颠倒混匀数次后,12000rpm离心10min;
⑥轻轻倒去上清液,在吸水纸上排干水分;
⑦加入300μl70%乙醇,轻轻颠倒混匀数次后,12000rpm离心2min;
⑧轻轻倒去上清液,在吸水纸上排干水分,置于37℃下15min,使乙醇充分挥发;
⑨用50μlddh2o重悬沉淀,得到基因组dna,浓度达到30ng/μl。
(3)转化子中抗潮霉素基因和gfp基因的pcr扩增
采用50μl反应体系进行pcr扩增,体系中含有:上下游引物各2μm,dntps200μm,mg2+1.5mm,10×pcrbuffer5μl,模版dna2μl,taq酶2u。
抗潮霉素基因的上游引物hph-f序列为5′-tcgttatgtttatcggcact-3′,下游引物hph-r序列为5′-gttcggtcggcatctact-3′。
gfp基因的上游引物gfp1序列为5′-ggcgagggcgagggcgatgc-3′,下游引物gfp2序列为5′-agttcaccttgatgccgttc-3′。
pcr扩增反应在朗基mg96g型pcr仪上进行。反应条件:94℃预变性2min,然后35个循环包括:94℃变性30sec,57℃退火40sec,72℃延伸1.5min。最后72℃后延伸10min。
(4)pcr产物的检测
pcr反应结束后,pcr产物经1%的琼脂糖凝胶电泳检测,结果发现这12个转化子中都出现了约850bp的抗潮霉素基因的电泳条带(图5),还出现了约400bp的gfp基因的的电泳条带(图6),说明抗潮霉素基因和gfp基因都稳定整合到葡萄座腔菌的基因组中。
(5)定量pcr检测转化基因的表达量
①转化子总rna的提取
菌丝体的培养方法同实施例2,用rnaisoplus来提取样本中的总rna,具体步骤如下:
1)将0.5g菌丝样品置于预冷的研钵中,在液氮中研磨成粉末。
2)加入1mlrnaisoplus,室温放置5min,使其充分裂解。
3)8500rpm离心5min,弃沉淀。
4)按每毫升rnaisoplus200μl氯仿的比例加入氯仿,混匀15s,室温放置15min,4℃8500rpm离心15min。
5)吸取上层水相,至另一个离心管中,按1:1体积加入异丙酮混匀,室温放置10min,4℃8500rpm离心10min,弃上清,rna沉于管底。
6)按每毫升rnaisoplus1ml75%乙醇的比例加入75%乙醇,轻轻振荡离心管,悬浮沉淀4℃,6500rpm离心5min,弃上清液,冰上晾干15min。
7)用100μl去除rna酶的h2o,溶解rna样品。
8)测od定量rna浓度,-70℃保存。
②cdna第一条链的合成
用反转录试剂盒primescriptrtreagentkitwithgdnaeraser((takara,dalianchina)合成第一条链。
1)总rna中基因组dna污染的除去反应
去除反应在冰上完成,按照表2制备去除混合液,低速离心混匀后,42℃反应2min,于4℃保存备用。
表2基因组dna去除反应混合液的制备
2)反转录反应
用takara反转录盒将rna反转录成cdna,具体步骤为:在冰上完成反转录反应混合液(表3)的制备,低速离心混匀后,37℃反应15min,85℃反应5s,4℃保存;用ddh2o将反应混合液稀释至200μl,-20℃保存。
表3反转率反应混合液的制备
③荧光定量pcr
定量pcr反应引物见表1
1)以tubulin基因作为内参,检测tubulin基因的引物对为bd-tubu/bd-tubd,检测抗潮霉素基因的引物对为q-hph-f/q-hph-r,检测gfp基因的引物对为gfp-f-rt/gfp-r-rt。引物合成由上海生工生物工程有限公司完成。
2)rt-pcr反应体系(25μl)
根据表4在冰上制备rt-pcr反应混合液,低速离心后,进行rt-pcr反应。
表4qrt-pcr反应混合液体系制备
qrt-pcr反应条件
qrt-pcr反应在icyceleriq荧光定量pcr仪(bio-rad,usa)上进行,反应采用两步法,反应条件设置为:95℃预变性30s;进入循环,95℃变性3s,55℃退火31s,共40个循环。
外源基因的相对表达量用公式e=2ctt-ctg计算,其中e是检测基因的相对表达量,ctt是tubulin基因表达的ct值,ctg是待检测基因的ct值。
本发明构建的载体pbdh3g-thy1和pbdh3g-thy2,以及用于对照的pko1-hph质粒转化葡萄座腔菌后,外源基因gfp和抗潮霉素基因hygromycinb(hph)都表达,本发明构建的载体的表达量,远远大于对照载体pko1-hph的表达量。其中抗潮霉素基因在pbdh3g-thy2的转化子中的表达量高于pko1-hph的11倍,而在pbdh3g-thy1中则超过108倍(图7)。gfp基因在pbdh3g-thy1和pbdh3g-thy2的转化子中的表达量高于pko1-hph的46倍(图8)。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!