一种快速体内验证基因打靶靶点效率的方法与流程
本发明属于基因组编辑技术领域,更具体地说是涉及一种快速体内验证基因打靶靶点效率的方法。
背景技术:
基因组编辑技术在最近几年快速发展,从最初的直接同源重组技术,到后来的基于切口的随机修复技术和基于切口的同源修复技术。现在,定点切口的产生是进行基因组编辑的关键,与之前的直接同源重组技术相比较,大大的提高了基因组编辑的效率。产生切口的位置也就是编辑的靶点,靶点的选择决定了基因组,因此定点产生切口的效率也就直接决定了基因组编辑的效率。在基于辛酯核酶,talen,crispr/cas9技术的基因组编辑中,都存在着靶点选择问题,选择一个高效的靶点就成了基因组编辑成功的前提条件。
技术实现要素:
本发明提出了一种快速体内验证基因打靶靶点效率的方法,本方法简单、快速,只需构建一次插入了靶点的载体,然后通过有颜色的菌斑计数的方式快速评价靶点效率。
一种快速体内验证基因打靶靶点效率的方法,基于细胞内的DNA断裂后会激发同源片段发生重组修复来实现的,通过重组使得报告基因的活性恢复的方式验证靶点的活性。
优选地,具体表现为:构建插入了靶点的载体,靶点断开后发生同源片段重组,将两个残缺的显色基因段重组成具有完整活性的蛋白,如果靶点具有活性将被切断,重组事件将被激活,显色基因被修复,从而产生有色菌斑;否则,重组事件不被激活,显色基因不被修复,然后通过显色基因产生的色斑计数的方式快速评价靶点效率。
优选地,靶点可以是任何类型的用于基因组DNA编辑的靶点。
优选地,报告基因是各种荧光蛋白或催化产生颜色反应的酶类的报告基因之一。
优选地,验证是在细胞内进行的,这些细胞可以是动物细胞,植物细胞或者细菌中的一种。
本发明产生的有益效果为:本方法构建一次插入了靶点的载体,然后通过有颜色的菌斑计数的方式快速评价靶点效率,本方法利用大肠杆菌中天然重组系统,同源片段发生重组,将两个残缺的显色基因段重组成具有完整活性的蛋白。如果靶点具有活性将被切断,重组事件将被激活,显色基因被修复,从而产生有色菌斑,否则,为正常颜色的大肠杆菌菌落。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
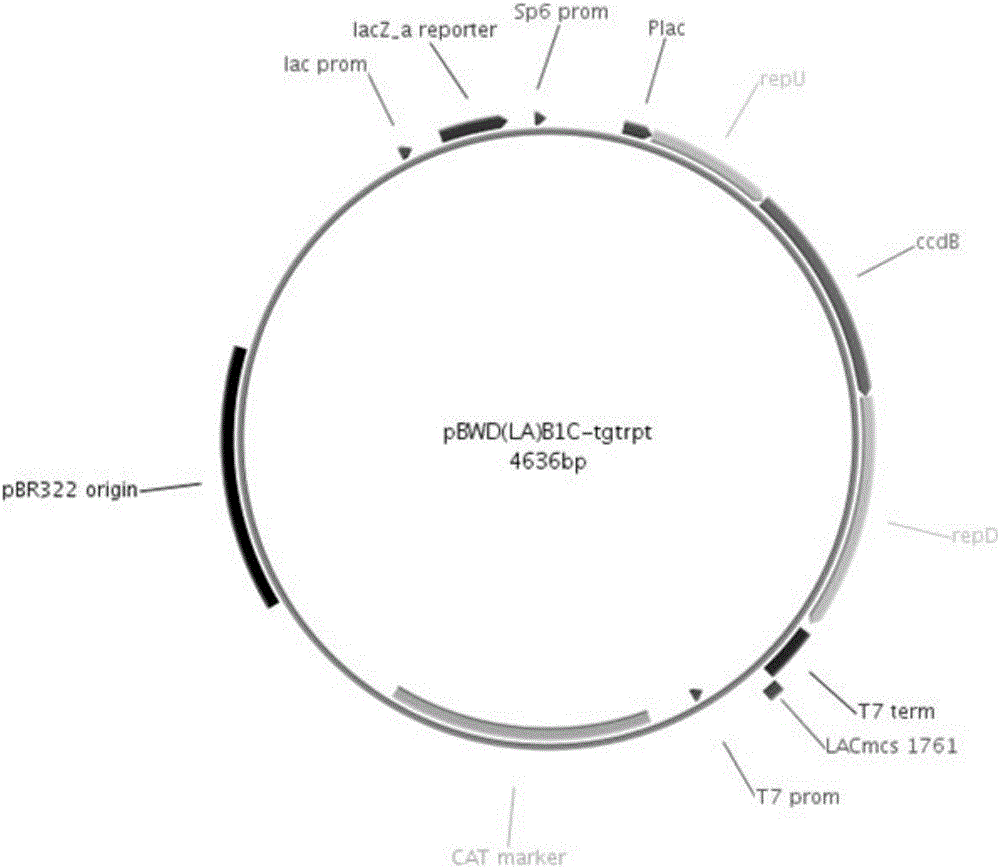
图1为载体pBWD(LA)BIC-tgtrpt构建结构图。
图2为载体PBWA(V)H-CAS9i构建结构图。
具体实施方式
实施例一:验证基于cas9/gRNA核酸酶的靶点效率
1、将靶标构建到靶点验证载体(载体1)中。
合成如下引物一对用于合成靶点序列(target:atttctttgaagcccaactt agg 23bp)。
Ptgt+:cagtGGTCTCaacatatttctttgaagcccaacttagg
Ptgt-:cagtGGTCTCa Gctg cctaagttgggcttcaaagaaa
===========PCR system=========
做1个50ul体系的PCR反应:
===========The PCR system&cycles===========
Total:50ul
Sum:1
H2O:34ul
buffer:5ul
Mg2+:4ul
dNTP:2ul
Ptgt+:2ul
Ptgt-:2ul
taq:2 2U
-----------pcr cycle-------------
94℃for 5min
+++++++++++30cycles
94℃for 30sec
50℃for 45sec
72℃for 1sec
+++++++++++++
72℃for 10min
16℃for 30min
将pcr产物与载体进行连接。
===========The Digesting-link Protocal===========
H2O:88ul
Buffer:22ul
BsaI/Eco31I:11ul
T4_ligase:11ul
pBWD(LA)BIC-tgtrpt:44ul
target:44ul
--------The Digest-Link(DL)procedure--------
37℃for 20min
++++++++5cycles
37℃for 10min
20℃for 10min
++++++++
37℃for 20min
80℃for 5min
将连接产物转化感受态
=========转化==========
将5-10ul连接产物转化大肠杆菌感受态(大肠杆菌感受态转化标准方法)
转化涂(氯霉素)抗性平皿,注意需要与v2载体不同抗性以便在共转化时用双抗筛选。,37℃培养12小时,进行菌斑pcr鉴定。
=============菌斑PCR鉴定========
挑取10个菌斑同时进行1.5mlEP管接菌和PCR鉴定,引物:pBWD(LA)B1C-tgtrpt;
鉴定引物Pmkt+:atgagtgtgagcaagggcgagga,Ptgt-.
==========PCR system==========[2015/11/28]
做10个25ul体系的PCR反应:
===========The PCR system&cycles===========
Total:25ul
Sum:10
H2O:16.5165ul
buffer:2.5 25ul
Mg2+:2 20ul
dNTP:1 10ul
pmkt+:1 10ul
Ptgt-:1 10ul
taq:1 10U
Template:1ul
-----------pcr cycle-------------
94℃for 5min
+++++++++++30cycles
94℃for 30sec
50℃for 45sec
72℃for 1sec
+++++++++++++
72℃for 10min
16℃for 30min
获得正确的克隆,进一步通过测序验证获得构建好的pBWD(LA)B1C-tgt鉴定载体(V1)。
2、将产生gRNA的序列构建到载体2中构建出(V2)。
合成如下1对引物
PgRNA+:cagtGGTCTCaggcatttctttgaagcccaactt
PgRNA-:cagtGGTCTCaaaacaagttgggcttcaaagaaa
用于合成gRNA:atttctttgaa gcccaactt
+=+=+=+=+=+
===========PCR system=========
做1个50ul体系的PCR反应:
===========The PCR system&cycles===========
Total:50ul
Sum:1
H2O:34ul
buffer:5ul
Mg2+:4ul
dNTP:2ul
PgRNA+:2ul
PgRNA-:2ul
taq:22U
-----------pcr cycle-------------
94℃for 5min
+++++++++++30cycles
94℃for 30sec
50℃for 45sec
72℃for 1sec
+++++++++++++
72℃for 10min
16℃for 30min
将pcr产物与载体进行连接。
===========The Digesting-link Protocal===========
H2O:8 8ul
Buffer:2 2ul
BsaI/Eco31I:1 1ul
T4_ligase:1 1ul
pBWA(V)H-cas9i:4 4ul
gRNA:4 4ul
--------The Digest-Link(DL)procedure--------
37℃for 20min
++++++++5cycles
37℃for 10min
20℃for 10min
++++++++
37℃for 20min
80℃for 5min
将连接产物转化感受态
=========转化==========
将5-10ul连接产物转化大肠杆菌感受态(见大肠杆菌感受态转化标准方法)
转化涂(卡纳霉素)抗性平皿,37℃培养12小时,进行菌斑pcr鉴定。
=============菌斑PCR鉴定========
挑取10个菌斑同时进行1.5mlEP管接菌和PCR鉴定,引物:pBWA(V)H-cas9i鉴定引物PgRNA+,Pbw2-(靶定在载体骨架上的序列Pbw2-:gcgattaagttgggtaacgccaggg).
==========PCR system==========
做10个25ul体系的PCR反应:
===========The PCR system&cycles===========
Total:25ul
Sum:10
H2O:16.5 165ul
buffer:2.5 25ul
Mg2+:2 20ul
dNTP:1 10ul
pgRNA+:1 10ul
Pbw2-:1 10ul
taq:1 10U
Template:1ul
-----------pcr cycle-------------
94℃for 5min
+++++++++++30cycles
94℃for 30sec
50℃for 45sec
72℃for 1sec
+++++++++++++
72℃for 10min
16℃for 30min
获得正确的克隆,进一步通过测序验证获得Cas9/gRNA表达载体(V2)。
3、共转化
1.在冰上解冻热击感受态细胞添加2μl DNA(V1,V2载体各1ul,)冰上培育约5分钟。
2.转移DNA/细胞混合物至42℃的温水中,保持90sec。
3.立刻放到冰上降温5min左右。
4.添加300ulz左右的LB,37℃下培养细胞30分钟至45min以复原(如果做质粒转化此步骤可以省略)。
5.转移细胞至氯霉素&卡纳霉素双抗培养基上培养。
第四歩:统计红斑数量
12-24小时候观察统计菌斑的红白斑比例,评估靶点效率。
共转化长出的红白菌斑,红斑代表靶点检测载体中,靶点被切并且红色荧光蛋白(mkate)重组修复。
不同靶点效率统计如下表:
实施例二:验证基于大肠杆菌的靶点效率
1、设计、构建靶点验证载体和作用于靶点的切割酶产生载体
如图1所示,载体pBWD(LA)BIC-tgtrpt骨架部分为pBR322载体,特别设计和构建部分为从Plac到T7term的部分。Plac为lac启动子,repU为报告基因(reporter)的上游序列,repD为报告基因(reporter)的下游序列,repU的下半部分和repD的上半部分是同源的序列,以便靶点断开后发生同源重组,将报告基因的完整序列复原,使其具有活性。ccdB为一般大肠杆菌的至死基因但是在菌株DB3.1中可以存活,因为起到负筛选作用,所以自链接的载体将会使被转化的一般大肠杆菌(DH5a,top10等)不能存活,从而提高靶标克隆的效率,T7term为T7终止子序列。构建时将靶标序列通过酶切链接的方式插入载体,并将CcdB(ccdb为常见大肠杆菌的至死蛋白,需在大肠杆菌DB3.1中才能够生存,分子克隆技术中常用来消除假阳性。)替换下来。
2、构建产生作用于靶点的蛋白或者其复合物的产生载体
A,如果是talen或者辛酯核酶技术,需要构建的是表达辛酯蛋白和talen内切酶的载体如图2,即为表达作用于靶标的蛋白后者蛋白复合物,构建方法与常规的表达载体构建相似,在此不做更多的表述。
B,如果是基于CAS9的技术,需要构建表达cas9蛋白和gRNA的载体,载体结构如图2。每次构建只需要将产生gRNA的19bp序列克隆进入载体代替ccdb序列即可。
3、将候选靶点克隆进靶点验证载体
将通过生物信息学分析好的候选靶点克隆到步骤1设计的靶点验证载体,代替ccdb序列,获得含有待验证靶点的载体。
4、将靶点验证载体与蛋白产生载体共转验证细胞
将三步获得的含有待验证靶点的载体与步骤2获得的能够表达作用于该靶点的蛋白的载体,一起共转化进入大肠杆菌。涂布平皿,经过过夜培养后,即可以统计菌斑数量以及有色菌斑比例,进而分析靶点的效率
5、统计比例评估靶点效率
统计有色菌斑的比例和颜色的亮度情况评估靶标的效率,红色菌斑越多越亮代表靶点效率越高,如果没有有色菌斑代表靶点无活性,不可作为后续转基因使用。
pBWD(LA)BIC-tgtrpt序列:
PBWA(V)H-CAS9i序列
- 还没有人留言评论。精彩留言会获得点赞!