NEP抑制剂及其药物组合物的制作方法
本发明属于药物
技术领域:
,具体涉及一种NEP抑制剂(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物及其药物组合物。
背景技术:
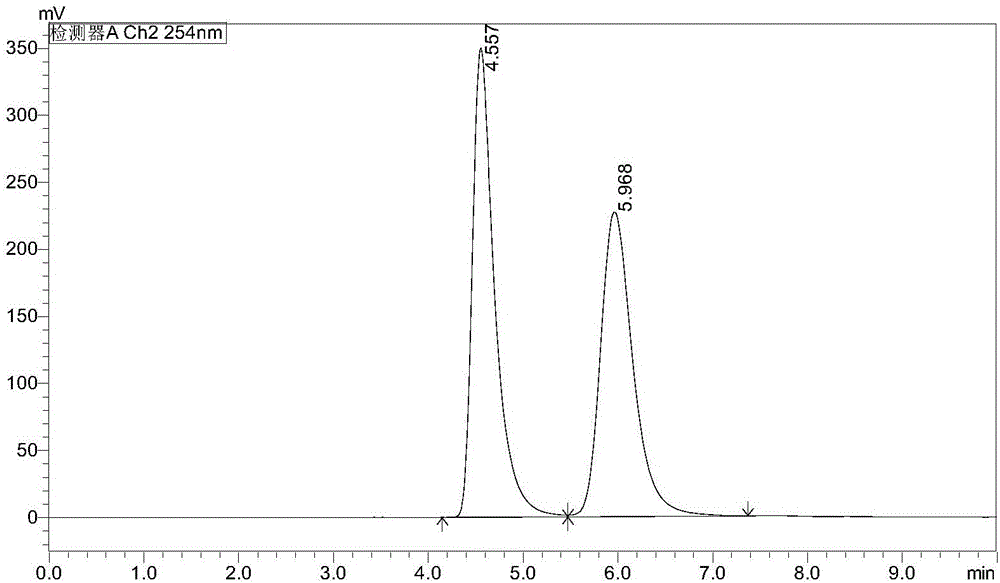
:高血压是最常见的心血管疾病之一,也是导致充血性心力衰竭、脑卒中、冠心病、肾功能衰竭、主动脉瘤的发病率和致死率升高的主要危险因素。据国际高血压学会最近发表的新闻公报,全球高血压或血压偏高人群已有9.72亿人,约占世界成年人口的26.4%。随着不断加剧的人口老龄化趋势,高血压病人的比例逐步上升。至2025年,估计患病人数将达到15.6亿。随着新一代抗高血压药物的相继问世和广泛应用,各类心血管疾病的死亡率有了较大幅度的下降。但是,全球每年仍有1700万人死于因高血压导致的心脑血管疾病,其中一半以上的病人死于急性心肌梗塞或脑血管栓塞症。2009年,中国治疗高血压药物的销售额已达到113.57亿元人民币,同比增长17.81%。目前我国高血压患者约有两亿人,每年还要新增高血压患者1000万。2006美国心脏学会估计美国心力衰竭人数接近580万;WorldFSSociety估计心力衰竭在西方的流行率为2.5%。我国成年人心衰的患病率为0.9%,患病人口约有400万。尽管经过多年的努力,但是我国高血压/心衰治疗率和控制率仍远远低于发达国家,因此我国此类药物市场发展前景广阔。NEP(neutralendopeptidase,又称Neprilysin)是M13锌依赖的金属蛋白酶家族的一种II型跨膜糖蛋白,也称脑啡肽酶,其上带有的锌原子处于酶的活性部位。NEP分子量约为97kDa,由750个氨基酸组成,包含一个信号肽和两个亲水结构域,胞内片段有26个氨基酸,胞外片段有700个氨基酸。NEP可作用于多肽的氨基端,使多肽链水解。自从1974年首次被定义以来,NEP作为一种神经肽的降解酶,已被发现广泛分布于内皮细胞、血管平滑肌细胞、心肌细胞、肾脏上皮细胞、纤维母细胞,其还存在于肺、肠、肾上腺、脑等,并具有众多组织特异性功能。钠利尿肽主要被NEP降解,而且NEP还能催化肾上腺髓质和缓激肽。NEP选择性抑制剂抑制NEP的活性,可以提高钠利尿肽及缓激肽的浓度,使血管扩张,心排出量增加,醛固酮水平下降并抑制RAAS(肾素-血管紧张素-醛固酮系统),降低心脏前、后负荷,延缓心肌重构,改善心力衰竭患者的心功能。近十年NEP的双重抑制剂的研究非常多,包括NEP/ACE双抑制剂、NEP/ARB(血管紧张肽受体)的双抑制剂、NEP/ECE(内皮素转化酶)的双抑制剂及NEP/APN(氨肽酶N)双抑制剂。NEP/ACE双抑制剂的抗高血压效应优于单一抑制剂,在于增加了P物质、缓激肽、肾上腺髓质素等舒张血管肽含量,减少了内皮素I、血管紧张素II等收缩血管肽含量,并优化结合了利尿和降压的双重效应,从而维持器官供血。Omapatrilat是BMS公司开发的强效ACE/NEP双抑制,但因为血管神经性水肿等副作用,2002年7月FDA拒绝批准Omapatrilat用于治疗高血压,甚至作为难治患者的最后治疗手段。Solvay公司研发有三个NEP/ECE(内皮素转化酶)的双抑制剂,目前处于临床二期阶段的daglutril(SLV-306,口服)和SLV-334(静脉给药)分别用于治疗肺动脉高压和脑损伤,处于临床一期阶段的SLV-338(静脉给药)用于治疗心血管疾病、代谢失调和神经性疾病。Pharmaleads公司研发的NEP/APN(氨肽酶N)双抑制剂PL-37(Debio-0827,口服)主要用于治疗疼痛,尤其是神经性疼痛。LCZ-696是由Novartis公司开发的口服降压药、保心药,是作用于NEP/ARB(血管紧张肽受体)的双抑制剂,目前处于临床三期阶段,最新的临床结果表明:在射血分数正常的心脏衰竭患者中,12周时LCZ-696引起的NT-proBNP降幅大于缬沙坦,且LCZ-696耐受性良好(NCT00887588)。至于这些效应能否转化为预后的改善,还需要进行前瞻性测试。Thomson-pharma预测其将于2014年上市,2017年的销售额将达到2.04亿美元。技术实现要素:本发明的目的在于解决目前存在的技术问题,提供一种(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物,这些化合物在体内都可以经代谢产生化合物LBQ657,具有NEP抑制活性,可以作为NEP抑制剂,可以用来治疗相关疾病,例如高血压、心衰、肺动脉高压以及肾脏疾病。本发明提供一种如式(Ⅰ)所示的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,其中,为R1、R2取代的手性碳原子;R1、R2各自独立的选自氢、氘、卤素、羟基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基,且R1、R2各不相同;R3选自氢、C1-8烷基、C3-8环烷基,任选进一步被一个或多个选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基、-S(O)rR4、-C(O)R4、-C(O)OR4、-O-C(O)R4、-O-C(O)OR4、-NR5R6、-C0-4-NR5R6、-C(O)NR5R6、-C0-4C(O)NR5R6或-N(R4)-C(O)R4取代基所取代,R4、R5、R6各自独立的选自氢、Cl-4烷基、C3-8环烷基,r为0、1、2。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R1、R2各自独立的选自氢、氘、卤素、羟基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C3-8环烷基,且R1、R2各不相同,R3、R4、R5、R6、r如式(Ⅰ)化合物所定义。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R1、R2各自独立的选自氢、氘、氟、甲基、乙基、异丙基、三氟甲基、羟甲基、环丙基、环已基、环丙氧基,且R1、R2各不相同,R3、R4、R5、R6、r如式(Ⅰ)化合物所定义。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R3选自氢、C1-4烷基、C3-6环烷基,任选进一步被一个或多个选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基取代基所取代,R1、R2如式(Ⅰ)化合物所定义。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R3选自氢、C1-4烷基、C3-6环烷基,任选进一步被一个或多个选自卤素、卤取代C1-8烷基、卤取代C1-8烷氧基、羟基、氰基、硝基、C1-8烷基取代基所取代,R1、R2如式(Ⅰ)化合物所定义。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R3选自氢、C1-4烷基、C3-6环烷基,任选进一步被一个或多个选自氟、三氟甲基、二氟甲基、羟基、氰基、硝基、二氟丙氧基、甲基、乙基、异丙基所取代,R1、R2如式(Ⅰ)化合物所定义。作为进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,R1,R2各自独立的选自氢、氟、甲基、乙基、异丙基、三氟甲基且R1、R2各不相同,R3选自氢、C1-4烷基、C3-6环烷基,任选进一步被一个或多个选自氟、三氟甲基、二氟甲基、二氟丙氧基、甲基、乙基、异丙基所取代。作为更进一步优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物,选自式(ⅠA)化合物或式(ⅠB)化合物:其中,R3选自C1-4烷基、C3-6环烷基。作为最优选的方案,所述的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物选自以下化合物:本发明的另一目的还在于提供一种制备式(I)所示的(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物的方法,包括对式(II)化合物进行手性拆分,R1、R2、R3如式(Ⅰ)化合物所定义;其中,手性拆分的HPLC流动相为:乙烷/异丙醇/醋酸,优选乙烷/异丙醇/醋酸的比例为70/30/0.1~80/20/0.1。手性拆分的HPLC检测波长为250nm~260nm,优选254nm。手性拆分的HPLC流速为0.5ml/min~5.0ml/min,优选1.0ml/min。手性拆分的HPLC进样量为1ul-10ul,优选2ul-8ul。本发明的再一目的还在于提供一种用于预防和/或治疗与NEP异常有关的疾病的药物组合物,所述药物组合物中含有治疗有效量的所述(2R,4S)-5-([1,1'-联苯基]-4-基)-4-(3-羧基丙酰氨基)-2-甲基戊酸酯衍生物、其立体异构体、水合物或溶剂合物。优选的,所述与NEP异常有关的疾病是指高血压、肺动脉高压、心衰以及肾脏疾病。本发明所述的系列化合物均具有明显的NEP抑制活性,经动物实验发现,本发明化合物具有良好的药代动力学活性,化合物溶解性能好,生物利用度高等特点,能够用于治疗或预防高血压、肺动脉高压、心衰以及肾脏疾病等与NEP异常有关的疾病的药物开发,有望开发出新一代NEP抑制剂。附图说明图1:实施例1化合物手性HPLC图谱(保留时间为4.163min的化合物登记为化合物(1-6R),保留时间为6.391的化合物登记为化合物(1-6S))。图2:实施例2化合物手性HPLC图谱(保留时间为4.557min的化合物登记为化合物(2-6R),保留时间为5.968min的化合物登记为化合物(2-6S))。具体实施方式详细说明:除非有相反陈述,下列用在说明书和权利要求书中的术语具有下述含义。“C1-8烷基”指包括1至8个碳原子的直链烷基和含支链烷基,烷基指饱和的脂族烃基团。例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基、正庚基、2-甲基己基、3-甲基己基、4-甲基己基、5-甲基己基、2,3-二甲基戊基、2,4-二甲基戊基、2,2-二甲基戊基、3,3-二甲基戊基、2-乙基戊基、3-乙基戊基、正辛基、2,3-二甲基己基、2,4-二甲基己基、2,5-二甲基己基、2,2-二甲基己基、3,3-二甲基己基、4,4-二甲基己基、2-乙基己基、3-乙基己基、4-乙基己基、2-甲基-2-乙基戊基、2-甲基-3-乙基戊基或其各种支链异构体等。烷基可以是取代的或未取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,优选为一个或多个以下基团,独立地选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基、-S(O)rR4、-C(O)R4、-C(O)OR4、-O-C(O)R4、-O-C(O)OR4、-NR5R6、-C0-4-NR5R6、-C(O)NR5R6、-C0-4C(O)NR5R6或-N(R4)-C(O)R4取代基所取代。“环烷基”指饱和或部分不饱和单环或多环环状烃取代基,“C3-8环烷基”指包括3至8个碳原子的环烷基,例如:单环环烷基的非限制性实施例包含环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等。多环环烷基包括螺环、稠环和桥环的环烷基。“螺环烷基”指单环之间共用一个碳原子(称螺原子)的多环基团,这些可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据环与环之间共用螺原子的数目将螺环烷基分为单螺环烷基、双螺环烷基基或多螺环烷基,螺环烷基的非限制性实施例包含:“稠环烷基”指系统中的每个环与体系中的其他环共享毗邻的一对碳原子的全碳多环基团,其中一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据组成环的数目可以分为双环、三环、四环或多环稠环烷基,稠环烷基的非限制性实施例包含:“桥环烷基”指任意两个环共用两个不直接连接的碳原子的全碳多环基团,这些可以含有一个或多个双键,但没有一个环具有完全共轭的π电子系统。根据组成环的数目可以分为双环、三环、四环或多环桥环烷基,桥环烷基的非限制性实施例包含:所述环烷基环可以稠合于芳基、杂芳基或杂环烷基环上,其中与母体结构连接在一起的环为环烷基,非限制性实施例包括茚满基、四氢萘基、苯并环庚烷基等。环烷基可以是任选取代的或未取代的,当被取代时,取代基优选为一个或多个以下基团,独立地选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基、-S(O)rR4、-C(O)R4、-C(O)OR4、-O-C(O)R4、-O-C(O)OR4、-NR5R6、-C0-4-NR5R6、-C(O)NR5R6、-C0-4C(O)NR5R6或-N(R4)-C(O)R4取代基所取代。“烷氧基”指-O-(烷基),其中烷基的定义如上所述。C1-8烷氧基指含1-8个碳的烷基氧基,非限制性实施例包含甲氧基、乙氧基、丙氧基、丁氧基等。烷氧基可以是任选取代的或未取代的,当被取代时,取代基优选为一个或多个以下基团,独立地选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基、-S(O)rR4、-C(O)R4、-C(O)OR4、-O-C(O)R4、-O-C(O)OR4、-NR5R6、-C0-4-NR5R6、-C(O)NR5R6、-C0-4C(O)NR5R6或-N(R4)-C(O)R4取代基所取代。“环烷氧基”指和-O-(未取代的环烷基),其中环烷基的定义如上所述。C3-8环烷氧基指含3-8个碳的环烷基氧基,非限制性实施例包含环丙氧基、环丁氧基、环戊氧基、环己氧基等。环烷氧基可以是任选取代的或未取代的,当被取代时,取代基优选为一个或多个以下基团,独立地选自卤素、羟基、氰基、硝基、C1-8烷基、卤取代C1-8烷基、羟基取代C1-8烷基、C1-8烷氧基、卤取代C1-8烷氧基、C3-8环烷基、C3-8环烷氧基、-S(O)rR4、-C(O)R4、-C(O)OR4、-O-C(O)R4、-O-C(O)OR4、-NR5R6、-C0-4-NR5R6、-C(O)NR5R6、-C0-4C(O)NR5R6或-N(R4)-C(O)R4取代基所取代。“卤素”指氟、氯、溴或碘。“-S(O)rR4”指R4取代的硫、亚硫酰基、硫酰基。“-C(O)R4”指R4取代的羰基。“-C(O)OR4”指R4取代的羰基氧基。“-O-C(O)R4”指R4取代的氧基羰基。“-O-C(O)OR4”指R4取代的氧基酯基。“-NR5R6”指R5、R6取代的氨基。“-C0-4-NR5R6”指R5、R6取代的C0-4烷基氨基。“-C(O)NR5R6”指R5、R6取代的酰氨基。“-C0-4C(O)NR5R6”指R5、R6取代的C0-4烷基酰氨基。“-N(R4)-C(O)R4”指R4取代氨基酰基。“任选”或“任选地”意味着随后所描述地事件或环境可以但不必发生,该说明包括该事件或环境发生或不发生地场合。例如,“任选被烷基取代的杂环基团”意味着烷基可以但不必须存在,该说明包括杂环基团被烷基取代的情形和杂环基团不被烷基取代的情形。“取代的”指基团中的一个或多个氢原子,优选为最多5个,更优选为1~3个氢原子彼此独立地被相应数目的取代基取代。不言而喻,取代基仅处在它们的可能的化学位置,本领域技术人员能够在不付出过多努力的情况下确定(通过实验或理论)可能或不可能的取代。例如,具有游离氢的氨基或羟基与具有不饱和(如烯属)键的碳原子结合时可能是不稳定的。“药物组合物”表示含有一种或多种本文所述化合物或其生理学上/可药用的盐或前体药物与其他化学组分的混合物,以及其他组分例如生理学/可药用的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。下面实施例将进一步举例说明本发明。这些实施例仅用于说明本发明,但不以任何方式限制本发明。实施例1第一步:1-氯乙基异丙基碳酸酯的制备化合物1-氯乙基氯甲酸酯(3ml,27.8mmol)用DCM(25ml)稀释,冷却至0℃,将用DCM(25ml)稀释的异丙醇(2.1ml,27.8mmol)用恒压滴液漏斗缓慢加入,反应15min左右,将用DCM(5ml)稀释的吡啶(2.4ml,30.6mmol)同样用恒压滴液漏斗缓慢加入,该过程中保持低温不变,加完后,室温下反应过夜,然后加水稀释,用DCM萃取,有机相干燥,浓缩后得3.74g淡黄色液体,产率为81%。1HNMR:(400MHz,CDCl3),6.36(q,1H),4.88(m,1H),1.759(dm,3H),1.26(t,6H)。第二步:1-碘乙基异丙基碳酸酯的制备化合物1-氯-乙基酯异丙基碳酸酯(3.74g,22.4mmol)溶解在乙腈(20ml)中,加入碘化钠(16.8g,112.2mmol),升温至50℃反应6小时左右,将溶剂旋干,固体用乙醚溶解,过滤掉多余的碘化钠,滤液旋干,得5.2g棕褐色液体,产率为90%。1HNMR:(400MHz,CDCl3),6.69(m,1H),4.87(m,1H),2.16(m,3H),1.25(m,6H)。第三步:(2R,4S)-4-(3-苄氧基羰基-丙酰氨基)-5-联苯基-4-基-2-甲基戊酸-1-异丙氧基碳氧基乙基酯的制备化合物(2R,4S)-4-(3-苄氧基羰基-丙酰氨基)-5-联苯基-4-基-2-甲基戊酸(284mg,0.6mmol)和Cs2CO3(391mg,1.2mmol)在乙腈中反应2.5小时后,将化合物1-碘-乙基异丙基碳酸酯(464mg,1.8mmol)加入,反应3小时后,补加化合物1-碘-乙基异丙基碳酸酯(464mg,1.8mmol)和Cs2CO3(391mg,1.2mmol),反应过夜,将体系旋干,反相柱层析纯化,得228mg产物。LC-MS:4.182min,[M+H]+:604,[M+Na]+:626。第四步:(2R,4S)-5-联苯基-4-基-4-(3-羧基-丙酰氨基)-2-甲基戊酸-1-异丙氧基碳氧基乙基酯的制备化合物(2R,4S)-4-(3-苄氧基羰基-丙酰氨基)-5-联苯基-4-基-2-甲基戊酸-1-异丙氧基碳氧基乙基酯(228mg)和Pd/C(0.2g)溶解在MeOH中,H2氛围下室温反应4小时左右,将Pd/C用硅藻土过滤掉,滤液旋干,反相柱层析纯化得到125mg淡黄色油状物(2R,4S)-5-联苯基-4-基-4-(3-羧基-丙酰氨基)-2-甲基戊酸-1-异丙氧基碳氧基乙基酯。1HNMR:(400MHz,CDCl3),7.49(m,2H),7.41(m,2H),7.35(m,2H),7.25(m,1H),7.13(m,2H),6.66(m,1H),4.82(m,1H),4.42(m,1H),3.20(m,1H),2.96(m,1H),2.4(m,6H),2.01(m,1H),1.69(m,1H),1.44(m,3H),1.23(m,6H),1.12(m,3H).LC-MS:4.045min,(M+Na)+:518。第五步:(6S,9R,11S)-11-([1,1'-联苯基]-4-基甲基)-2,6,9-三甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸(1-6S)和(6R,9R,11S)-11-([1,1'-联苯基]-4-基甲基)-2,6,9-三甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸(1-6R)的制备化合物(1-6S)和化合物(1-6R)是由化合物(6)经手性柱拆分得到。化合物手性拆分采用HPLC法,使用手性制备仪器和合适的手性柱(大赛璐公司)对手性异构体进行分离,收集其相应组分。旋转蒸发除去溶剂,得到光学异构体的纯品。手性HPLC图谱见附图1,柱效能报告见下表:峰号保留时间面积面积%T塔板数拖尾因子分离度14.163657639044.55926091.188--25.3251102560.74739960.9583.50636.391797053154.00524751.1352.50448.7491016560.68960541.2914.896手性拆分条件如下:化合物核磁数据如下:1、化合物(1-6S):1HNMR(400MHz,CDCl3)δ7.55-7.48(m,2H),7.46(d,J=8.1Hz,2H),7.36(t,J=7.5Hz,2H),7.26(t,J=7.3Hz,1H),7.16(d,J=8.1Hz,2H),6.52–6.42(m,2H),4.81(hept,J=6.2Hz,1H),4.33(brs,1H),2.77(dd,J=13.6,6.6Hz,1H),2.72–2.47(m,4H),2.44–2.38(m,2H),1.87(ddd,J=14.3,7.6,3.7Hz,1H),1.81–1.69(m,1H),1.46(d,J=5.3Hz,3H),1.26(d,J=6.2Hz,3H),1.24(d,J=6.2Hz,3H),1.07(d,J=7.0Hz,3H);LC-MS:tR4.31min,[M+H]+:514.1,[M+Na]+:536.2。2、化合物(1-6R):1HNMR(400MHz,CDCl3)δ7.55-7.49(m,2H),7.46(d,J=8.0Hz,2H),7.36(t,J=7.6Hz,2H),7.26(t,J=7.3Hz,1H),7.17(d,J=8.0Hz,2H),6.68(q,J=5.4Hz,1H),5.69(d,J=7.0Hz,1H),4.86–4.74(m,1H),4.20(brs,1H),2.79(d,J=6.0Hz,3H),2.68–2.24(m,9H),1.87(ddd,J=13.5,9.6,4.0Hz,1H),1.55(ddd,J=13.7,10.5,3.5Hz,1H),1.45(d,J=5.4Hz,3H),1.23(d,J=2.1Hz,3H),1.22(d,J=2.1Hz,4H),1.11(d,J=7.1Hz,3H);LC-MS:tR4.28min,[M+H]+:514.1,[M+Na]+:536.2。实施例2第一步:(9R,11S)-11-([1,1'-联苯基]-4-基甲基)-6-异丙基-2,9-二甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸的制备化合物(2R,4S)-4-(3-苄氧基羰基-丙酰氨基)-5-联苯基-4-基-2-甲基戊酸(1410mg,2.98mmol)溶于10mLN,N-二甲基甲酰胺,加入Cs2CO3(580mg,1.79mmol),室温搅拌1小时,加入KI(1240mg,7.44mmol),1-氯-2-甲基丙基异丙基碳酸酯(1450mg,7.44mmol),加热65度反应24小时。反相柱层析分离得745mg粗产品(含合环副产物)。再将其溶于10mL甲醇,加入80mg钯/碳,常温常压氢化5小时。过滤,浓缩,反相柱层析分离,得白色泡状固体(9R,11S)-11-([1,1'-联苯基]-4-基甲基)-6-异丙基-2,9-二甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸(534mg,两步33%)。LC-MS:tR:3.11min,[M+H]+:542.2,[M+Na]+:564.3。第二步:(6S,9R,11S)-11-([1,1'-联苯基]-4-基甲基)-6-异丙基-2,9-二甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸(2-6S)和(6R,9R,11S)-11-([1,1'-联苯基]-4-基甲基)-6-异丙基-2,9-二甲基-4,8,13-三羰基-3,5,7-三氧杂-12-氮杂十六烷-16-酸(2-6R)的制备化合物(2-6S)和化合物(2-6R)是由化合物(9)经手性柱拆分得到。化合物手性拆分采用HPLC法,使用手性制备仪器和合适的手性柱(大赛璐公司)对手性异构体进行分离,收集其相应组分。旋转蒸发除去溶剂,得到光学异构体的纯品。手性HPLC图谱见附图2,柱效能报告见下表:峰号保留时间面积面积%拖尾因子(10%)分离度高度%14.557min4.557574143251.4891.543--25.968min5.968540925348.5111.2672.781手性拆分条件如下:化合物核磁数据如下:1、化合物(2-6S):1HNMR(400MHz,CDCl3)δ7.57(d,J=7.4Hz,2H),7.52(d,J=7.7Hz,2H),7.43(t,J=7.5Hz,2H),7.33(t,J=7.3Hz,1H),7.23(d,J=7.8Hz,2H),6.48–6.38(m,2H),4.87(dt,J=12.4,6.1Hz,1H),4.36(brs,1H),2.87(dd,J=13.2,5.9Hz,1H),2.80–2.38(m,6H),2.15–2.04(m,1H),3.02–1.90(m,1H),1.87–1.72(m,1H),1.31(t,J=6.8Hz,6H),1.16(d,J=6.8Hz,3H),0.98(d,J=6.8Hz,6H)。2、化合物(2-6R):1HNMR(400MHz,CDCl3)δ7.57(d,J=7.3Hz,2H),7.52(d,J=7.9Hz,2H),7.43(t,J=7.6Hz,2H),7.34(d,J=7.3Hz,1H),7.23(d,J=8.0Hz,2H),6.50(d,J=4.8Hz,1H),5.78(d,J=7.6Hz,1H),4.94–4.80(m,1H),4.26(brs,1H),2.96–2.79(m,2H),2.63(s,3H),2.46(s,2H),2.12–2.00(m,1H),1.99–1.89(m,1H),1.64(t,J=10.5Hz,1H),1.32–1.26(m,6H),1.18(d,J=7.0Hz,3H),1.02–0.95(m,6H)。生物学实验例1(大鼠药代动力学研究试验)1.研究目的以大鼠为受试动物,研究NEP系列前药化合物(6),化合物(1-6R),化合物(2-6R),化合物(2-6S)在大鼠体内的药代动力学行为,评价其药动学特征。2.试验方案2.1试验动物每例使用SD大鼠3只,雄性,由西普尔-必凯实验动物有限公司提供,动物生产许可证号SCXK(沪)2008-0016。2.2药物配制样品全取(按30mg计),加1ml乙醇使溶解,后依次加入1ml(温水适当加热使融化)SolutolHS15和8mlpH4.63醋酸盐缓冲液,制成3mg/ml溶液,剩余药液留样用于定量分析。2.3给药SD大鼠3只,雄性;禁食一夜后分别灌胃给药,剂量为30mg/kg,给药体积10ml/kg。2.4样品采集于给药前和给药后0.5,1.0,2.0,4.0,6.0,8.0,24.0h采血0.1ml,置于肝素化试管中,4℃3500rpm离心10min分离血浆,于-20℃保存;给药后2h进食。因待测物在血浆中不稳定,血样采集后迅速置于冰水浴中,血样采集及处理、分析的全过程保持低温状态。3.分析方法3.1仪器设备API4000三重四级杆质谱仪,美国AppliedBiosystems公司;ShimadzuLC-20AD高效液相色谱系统,日本Shimadzu公司。3.2色谱条件色谱柱:InertsilC8-3(50×4.6mm,5μm);流动相:甲醇:0.2%甲酸水溶液(梯度洗脱);流速(ml/min):1.0ml/min。3.3质谱条件3.4血浆样品预处理同时检测前药及代谢产物LBQ657:取给药后各时刻的大鼠血浆25ul,加入内标SHR133162(200ng/ml,甲醇配制)50ul,甲醇175ul,涡旋混合3min,离心10min(13500rpm),取上清液10ul进行LC/MS/MS分析。3.5标准曲线制备同时检测前药及代谢产物LBQ657:取大鼠空白血浆25ul,加入混合标准系列溶液25ul,使血药浓度为1.00,2.00,5.00,25.0,100,500,2000,5000,20000和40000ng/ml,加入内标SHR133162(200ng/ml,甲醇配制)50ul,甲醇150ul,按“血浆样品预处理”项下操作。以血药浓度为横坐标,样品与内标色谱峰面积比为纵坐标,以加权最小二乘法(w=1/x2)进行线性回归,获得典型标准曲线方程如下表:4.NEP化合物大鼠药代动力学研究1.大鼠灌胃给予30mg/kg前药化合物(6)后,各时刻化合物(6)血药浓度均低于定量下限,推测该前药在大鼠体内快速被酶水解;代谢产物LBQ657的血药浓度达峰时间tmax为0.83h,峰浓度Cmax为36393ng/ml,血药浓度-时间曲线下面积AUC0-t为54682ng/ml·h,消除半衰期t1/2为2.29h。2.大鼠灌胃给予30mg/kg前药化合物(1-6R)后,化合物(1-6R)在大鼠体内各时刻血药浓度均低于定量下限,推测该前药被酶快速水解;代谢产物LBQ657的血药浓度达峰时间tmax为0.43h,峰浓度Cmax为15056ng/ml,血药浓度-时间曲线下面积AUC0-t为33505ng/ml·h,消除半衰期t1/2为1.91h。3.大鼠灌胃给予30mg/kg前药化合物(2-6R)后,化合物(2-6R)在大鼠体内各时刻血药浓度均低于定量下限,推测该前药被酶快速水解;代谢产物LBQ657的血药浓度达峰时间tmax为1.0h,峰浓度Cmax为6120ng/ml,血药浓度-时间曲线下面积AUC0-t为10713ng/ml·h,消除半衰期t1/2为0.98h。4.大鼠灌胃给予30mg/kg前药化合物(2-6S)后,化合物(2-6S)在大鼠体内各时刻血药浓度均低于定量下限,推测该前药被酶快速水解;代谢产物LBQ657的血药浓度达峰时间tmax为0.5h,峰浓度Cmax为6140ng/ml,血药浓度-时间曲线下面积AUC0-t为10989ng/ml·h,消除半衰期t1/2为1.2h。结论:本发明实施例化合物均可以在大鼠体内快速地代谢成活性药物成分LBQ657。生物学实验例2(犬药代动力学研究试验)1.研究目的以比格犬为受试动物,研究NEP系列前药化合物(6),化合物(1-6R),化合物(1-6S),(2-6R),(2-6S)在比格犬体内的药代动力学行为,评价其药动学特征。2.试验方案2.1试验动物每例使用3只Beagle犬,由苏州西山中科实验动物有限公司提供。2.2药物配制称取样品,用乙醇,SolutolHS15和pH4.63醋酸盐缓冲液(1:1:8),制成1mg/ml溶液,剩余药液留样用于定量分析。2.3给药禁食一夜后分别灌胃给药,剂量为3mg/kg。2.4样品采集于给药前和给药后0.5,1.0,2.0,4.0,6.0,8.0,24.0h采血0.1ml,置于肝素化试管中,4℃3500rpm离心10min分离血浆,于-20℃保存;给药后3h进食。因待测物在血浆中不稳定,血样采集后迅速置于冰水浴中,血样采集及处理、分析的全过程保持低温状态。3.分析方法3.1仪器设备API4000三重四级杆质谱仪,美国AppliedBiosystems公司;ShimadzuLC-20AD高效液相色谱系统,日本Shimadzu公司。3.2色谱条件色谱柱:InertsilC8-3(50×4.6mm,5μm);流动相:甲醇:0.2%甲酸水溶液(梯度洗脱);流速(ml/min):1.0ml/min。3.3质谱条件3.4血浆样品预处理同时检测前药及代谢产物LBQ657:取给药后各时刻的大鼠血浆25ul,加入内标SHR133162(200ng/ml,甲醇配制)50ul,甲醇175ul,涡旋混合3min,离心10min(13500rpm),取上清液10ul进行LC/MS/MS分析。3.5标准曲线制备同时检测前药及代谢产物LBQ657:取大鼠空白血浆25ul,加入混合标准系列溶液25ul,使血药浓度为1.00,2.00,5.00,25.0,100,500,2000,5000,20000和40000ng/ml,加入内标SHR133162(200ng/ml,甲醇配制)50ul,甲醇150ul,按“血浆样品预处理”项下操作。以血药浓度为横坐标,样品与内标色谱峰面积比为纵坐标,以加权最小二乘法(w=1/x2)进行线性回归,获得典型标准曲线方程如下表:4.NEP化合物比格犬药代动力学研究1.犬灌胃给予3.0mg/kg化合物(6)后,平均血药浓度达峰时间tmax为0.50h,平均峰浓度Cmax为462ng/ml,平均血药浓度-时间曲线下面积AUC0-t为514ng/ml·h;其代谢物LBQ657的平均血药浓度达峰时间tmax为0.75h,平均峰浓度Cmax为294ng/ml,平均血药浓度-时间曲线下面积AUC0-t为428ng/ml·h,平均消除半衰期t1/2为2.21h。2.犬灌胃给予3.0mg/kg化合物(1-6R)后,平均血药浓度达峰时间tmax为0.50h,平均峰浓度Cmax为648ng/ml,平均血药浓度-时间曲线下面积AUC0-t为598ng/ml·h;其代谢物LBQ657的平均血药浓度达峰时间tmax为0.50h,平均峰浓度Cmax为627ng/ml,平均血药浓度-时间曲线下面积AUC0-t为762ng/ml·h,平均消除半衰期t1/2为1.75h。3.犬灌胃给予3.0mg/kg化合物(1-6S)后,化合物例3在犬体内各时刻血药浓度均低于定量下限,推测该前药被酶快速水解;其代谢物LBQ657的平均血药浓度达峰时间tmax为0.50h,平均峰浓度Cmax为542ng/ml,平均血药浓度-时间曲线下面积AUC0-t为671ng/ml·h,平均消除半衰期t1/2为1.64h。4.犬灌胃给予3.0mg/kg化合物(2-6R)后,平均血药浓度达峰时间tmax为0.08h,平均峰浓度Cmax为611ng/ml,平均血药浓度-时间曲线下面积AUC0-t为134ng/ml·h;其代谢物LBQ657的平均血药浓度达峰时间tmax为0.25h,平均峰浓度Cmax为223ng/ml,平均血药浓度-时间曲线下面积AUC0-t为150ng/ml·h,平均消除半衰期t1/2为2.91h。5.犬灌胃给予3.0mg/kg化合物(2-6S)后,平均血药浓度达峰时间tmax为0.31h,平均峰浓度Cmax为829ng/ml,平均血药浓度-时间曲线下面积AUC0-t为831ng/ml·h;其代谢物LBQ657的平均血药浓度达峰时间tmax为0.50h,平均峰浓度Cmax为136ng/ml,平均血药浓度-时间曲线下面积AUC0-t为168ng/ml·h,平均消除半衰期t1/2为2.35h。结论:本发明实施例化合物均可以在犬体内代谢成活性药物成分LBQ657。生物学实验例3(NEP酶抑制测试)下面的体外试验可用来测定本发明化合物对于大鼠血浆中NEP酶的抑制活性,其活性可用IC50值来表示。化合物的半数抑制浓度IC50(将一定浓度的酶活性抑制至50%时所需的化合物浓度)是通过将一定量的酶与特定的底物及不同浓度的待测化合物混合反应后测定计算出的。本实验所用的NEP酶体系是取自正常SD大鼠的新鲜血液,置于含肝素钠抗凝剂的管中,5000rpm,4℃离心10min后,收集血浆;配制不同浓度的NEP抑制剂,其中,NEP抑制剂的最高终浓度为100μM,并以3倍梯度依次稀释10次;每孔加60μl新鲜的大鼠血浆于384孔板;随后,每孔再加入10μl梯度稀释的NEP抑制剂;37℃,孵育30min后,每孔加入10μl10μM的底物(SenoLyte520NeprilysinActivityAssayKit,AnaSpec,72223);在37℃孵育18h(±1h);在激发光490nm和发射光520nm的波长下,测定荧光信号;并采用Prism5.0软件求得IC50。本发明化合物的NEP酶抑制活性通过以上的试验进行测定,测得的IC50值见下表。化合物IC50(uM)化合物(1-6S)0.035化合物(1-6R)0.038化合物(2-6S)0.040化合物(2-6R)0.042结论:本发明化合物在SD大鼠血浆中对NEP酶均有明显地抑制作用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1