作为用于治疗癫痫、成瘾和肝细胞癌的GABA氨基转移酶灭活剂的(S)-3-氨基-4-(二氟亚甲基)环戊-1-烯-1-羧酸及有关化合物的制作方法
本发明在由美国国立卫生研究院(National Institute of Health)授予的R01 DA030604的政府支持下进行。政府在本发明中具有一定权利。
发明背景
γ-氨基丁酸(GABA),中枢神经系统中的主要抑制性神经递质,从突触前抑制性神经元释放并结合至氯化物选择性离子通道受体(GABAA和GABAC)并且结合至G-蛋白偶联受体(GABAB)以使突触后膜超极化,从而向下地控制神经元活性。低水平的GABA与许多神经紊乱有关,包括癫痫、帕金森病、阿尔兹海默病、亨廷顿氏病以及可卡因成瘾。
γ-氨基丁酸能药物是改进GABA的分泌和传递的药物。这些药物作为家族已经被用于治疗许多神经系统紊乱,所述神经系统紊乱包括纤维肌痛(fibromyalgia)、神经病变、癫痫有关偏头痛(migraines related to epilepsy)、不宁腿综合征(restless leg syndrome)和创伤后应激障碍。γ-氨基丁酸能药物包括GABAA和GABAB受体配体、GABA再摄取抑制剂、GABA氨基转移酶抑制剂、GABA类似物或包含GABA自身的分子。
在1998年,开发了用于通过抑制γ-氨基丁酸氨基转移酶(GABA-AT)的活性来治疗可卡因成瘾的新颖的策略,γ-氨基丁酸氨基转移酶是降解GABA的5'-磷酸吡多醛(pyridoxal 5'-phosphate)(PLP)依赖性酶。GABA-AT抑制升高GABA水平,这拮抗伏隔核(nucleus accumbens)(NAcc)中的多巴胺的快速释放,多巴胺是响应于可卡因滥用和其他药物滥用的神经化学物质。从那以后,当前用作抗癫痫药物的唯一FDA批准的GABA-AT的灭活剂氨己烯酸一直在动物模型中用于可卡因、尼古丁、甲基苯丙胺、海洛因以及酒精的成瘾的治疗方面是成功的。氨己烯酸还在治疗人类中的可卡因成瘾方面是有效的,其中多达28%的患者在9周的双盲试验中实现戒瘾。然而,用于一般治疗用途的氨己烯酸的潜力可能是有问题的,因为已经报告,在25%-40%的癫痫患者中,永久性视力损失(permanent vision loss)由氨己烯酸的长期施用引起。
最近,现在被称为CPP-115的(1S,3S)-3-氨基-4-二氟亚甲基-1-环戊酸((1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid)被设计、合成并且发现在灭活GABA-AT方面比氨己烯酸更有效186倍。(Pan,Y.;Qiu,J.;Silverman,R.B.J.Med.Chem.2003,46(25),5292-5293)。当在婴儿痉挛症(infantile spasm)的多击大鼠模型(multiple-hit rat model)中测试时,CPP-115以比在氨己烯酸的情况下使用的剂量低>100倍的剂量抑制痉挛症。CPP-115还具有比氨己烯酸大得多的安全系数和相当低的视网膜毒性易感性(retinal toxicity liability)。当在施用20mg/kg可卡因之后自由运动大鼠中测试时,在减少NAcc中的多巴胺的释放方面,CPP-115比氨己烯酸更有效>300倍。(Silverman,R.B.J.Med.Chem.2012,55(2),567-575;Pan,Y.;Gerasimov,M.R.;Kvist,T.;Wellendorph,P.;Madsen,K.K.;Pera,E.;Lee,H.;Schousboe,A.;Chebib,M.;-Osborne,H.;Craft,C.M.;Brodie,J.D.;Schiffer,W.K.;Dewey,S.L.;Miller,S.R.;Silverman,R.B.J.Med.Chem.2012,55(1),357-366)。另外,将以1mg/kg的CPP-115连同可卡因施用至可卡因成瘾的大鼠,在消除其成瘾行为方面示出与以300mg/kg的氨己烯酸连同可卡因类似的效果。
初始地,CPP-115被设计成经由将导致与酶的共价加合物的迈克尔加成机制灭活GABA-AT。然而,后来发现,CPP-115通过经由在产生的代谢物中的两个羧酸盐基团与活性位点中的Arg192和Arg445的强的静电相互作用与酶形成紧密结合的复合物来灭活该酶(方案1)。(Lee,H.;Doud,E.H.;Wu,R.;Sanishvili,R.;Juncosa,J.I;Liu,D.;Kelleher,N.L.;Silverman,R.B.J.Am.Chem.Soc.2015,137(7),2628-2640)。代谢通过CPP-115与赖氨酸结合的PLP形成席夫碱来引发,随后是γ-质子除去和互变异构化,产生迈克尔受体。然而,在Lys-329可以攻击此迈克尔受体之前,发生二氟亚甲基基团的催化水解,产生PLP-结合的二羧酸盐代谢物,所述二羧酸盐代谢物经历构象变化并紧密结合至Arg192和Arg445(方案1)。然而,分子建模指示在互变异构化之后二氟亚甲基基团的弯曲远离Lys-329的运动,这使其对于亲核攻击太远(方案1)。相反地,推测起来酶催化二氟亚甲基基团的水解。
方案1.通过CPP-115灭活GABA-AT的机制
发明概述
鉴于前述,本发明的目的是提供用于选择性抑制GABA-AT,从而克服包括上文概述的那些的现有技术的各种缺陷和缺点的化合物、组合物和有关方法。本领域技术人员将理解的是,本发明的一个或更多个方面可以满足某些目的,同时一个或更多个其他方面可以满足某些其他目的。在其所有方面中每个目的可能不同样地适用于本发明的每一个方面。因此,以下目的可以在关于本发明的任一方面的可选方案中被观察到。
本发明的目的可以是提供呈现出选择性氨基转移酶抑制的一种或更多种小分子、非肽化合物。
本发明的另一个目的可以是提供用于在指示一种或更多种哺乳动物疾病状态的条件下体外使用和研究的一种或更多种此类化合物。
可选择地,本发明的目的还可以是提供能够体内治疗此类疾病状态的一种或更多种此类化合物。
本发明的目的还可以是单独地或与前述目的中的一个或更多个联合地提供用于成瘾和相关指征的GABA-AT灭活、抑制或调节和/或治疗的化合物或组合物。
本发明的目的还可以是单独地或与前述目的中的一个或更多个联合地提供用于恶性病理性增殖紊乱(malignant pathologic proliferative disorder)的OAT灭活、抑制或调节和/或治疗的化合物或组合物,所述恶性病理性增殖紊乱包括而不限于肝细胞癌。
本发明的目的还可以是单独地或与前述目的中的一个或更多个联合地提供用于一系列神经紊乱或心理紊乱的治疗的化合物或组合物,所述神经或心理紊乱包括但不限于本文别处描述的那些。
本发明的其他目的、特征、益处和优点将从此概述和此类化合物、组合物和/或方法的某些实施方案的以下描述中是明显的,并且将对于具有本文描述的合成技术的知识的本领域技术人员是容易地明显的。当结合随附的实施例、数据、附图和本文并入的参考文献连同将从其中得到的所有合理的推断时,这样的目的、特征、益处和优点将从上文是明显的。
部分地,本发明可以涉及下式的化合物:
其中R1和R2可以独立地选自H、F、Cl、Br以及I,其中R1和R2中的至少一个不是H;或此类化合物的盐。不受限制地,在某些实施方案中,包含氨基取代基的立构中心可以具有(S)立体化学构型。
部分地,本发明可以涉及下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F;或此类化合物的盐。在某些实施方案中,R1和R2可以是F。不受限制地,在某些此类实施方案中,包含氨基取代基的立构中心可以具有(S)立体化学构型。
部分地,本发明可以涉及下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F,或此类化合物的盐。在某些非限制性实施方案中,R1和R2可以是F。
无论如何,本发明的化合物或联合本发明可用的化合物没有立体化学限制或构型限制。如下文说明和讨论的,此类化合物和/或其中间体作为单个对映异构体或作为异构体可以从其中拆分的外消旋混合物是可用的。因此,任何立构中心可以是(S)或(R)。作为单独的考虑,关于单取代的亚甲基实施方案,此类化合物可以具有Z构型或E构型。作为另一种单独的考虑,多种化合物可以作为部分地或完全地质子化的酸式盐存在。在某些此类实施方案中,关于铵基取代基,反荷离子可以是质子酸的共轭碱。在某些此类或其他实施方案中,关于羧酸盐取代基,反荷离子可以是碱阳离子、碱土阳离子或铵阳离子。另外,本领域技术人员将理解,本发明的任一种或更多种化合物可以作为用于与治疗方法或药物联合使用的包含药学上可接受的载体组分的药物组合物的部分来提供。
部分地,本发明可以涉及降低、抑制、调节或以其他方式影响GABA-AT活性的方法。此类方法可以包括提供下式的化合物:
其中R1和R2可以独立地选自H、F、Cl、Br以及I,其中R1和R2中的至少一个不是H,或此类化合物的盐;以及使此类化合物与包含γ-氨基丁酸氨基转移酶的介质(medium)接触,如可能的此类化合物是以足以降低、抑制、调节或以其他方式影响这样的氨基转移酶活性的量。此类方法从而可以将此类化合物结合至此类氨基转移酶和/或灭活此类氨基转移酶,并且升高或调节此类介质中的γ-氨基丁酸水平。这样的接触可以是在体外或在体内。可选择地,本发明可以被认为是用于治疗在需要其的受试者中的低水平γ-氨基丁酸的方法。无论如何,在某些非限制性实施方案中,R1和R2可以是F。
部分地,本发明还可以涉及抑制、调节、阻止或以其他方式影响响应于成瘾物质的摄取的多巴胺的释放或升高的方法。此类方法可以包括提供下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F,或此类化合物的盐;以及使此类化合物与包含γ-氨基丁酸氨基转移酶的细胞介质(cellular medium)接触,此类化合物是以足以调节或抑制响应于这样的成瘾物质的摄取或响应于成瘾行为的多巴胺水平的量。此类方法从而可以增加γ-氨基丁酸水平并调节和/或控制多巴胺水平。可选择地,本发明可以被认为是用于治疗受成瘾挑战和/或以其他方式需要其的受试者中的过量多巴胺释放的方法。无论如何,在某些非限制性实施方案中,R1和R2可以是F。
部分地,本发明还可以涉及用于治疗在需要其的哺乳动物受试者中的物质成瘾的方法,所述物质成瘾例如并且不限于可卡因成瘾、海洛因成瘾、酒精成瘾、巴比妥酸盐成瘾、安非他命成瘾、大麻成瘾、美沙酮成瘾、阿片样物质成瘾、兴奋剂成瘾以及尼古丁成瘾及其组合。此类方法可以包括向此类受试者施用下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F,或此类化合物的盐,如可能的此类化合物是以足以在已经摄取例如可卡因、海洛因、酒精、巴比妥酸盐、安非他命、大麻、美沙酮、阿片样物质、兴奋剂以及尼古丁及其组合的受试者的海马体(hippocampus)中增加γ-氨基丁酸水平和/或调节/控制的多巴胺水平的量。此类方法从而可以降低海马体葡萄糖代谢。在某些非限制性实施方案中,R1和R2可以是F。
部分地,本发明可以涉及降低、抑制、调节或以其他方式影响OAT活性的方法。此类方法可以包括提供下式的化合物:
其中R1和R2可以独立地选自H、F、Cl、Br以及I,其中R1和R2中的至少一个不是H,或此类化合物的盐;以及使此类化合物与包含鸟氨酸氨基转移酶的介质接触,如可能的此类化合物是以足以降低、抑制、调节或以其他方式影响这样的氨基转移酶活性的量。这样的接触可以是在体外或在体内。无论如何,在某些非限制性实施方案中,R1和R2可以是F。
部分地,本发明还可以涉及降低通过人类肝细胞癌表达的鸟氨酸氨基转移酶的活性的方法。此类方法可以包括提供下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F,或此类化合物的盐;以及使此类化合物与包含表达鸟氨酸氨基转移酶的肝细胞癌的细胞介质接触,如可能的此类化合物是以足以调节或降低鸟氨酸氨基转移酶活性的量。此类方法从而可以降低此类介质中的谷氨酸产生。无论如何,在某些非限制性实施方案中,R1和R2可以是F。
部分地,本发明还可以涉及用于治疗心理紊乱或神经紊乱的方法。此类方法可以包括向需要其的哺乳动物受试者施用下式的化合物:
其中R1和R2可以选自H和F,并且R1和R2中的至少一个可以是F,或此类化合物的盐,如可能的此类化合物是以足以增加此类受试者中的γ-氨基丁酸水平的量。不受限制地,心理紊乱和神经紊乱可以选自本文别处讨论的那些。无论如何,在某些非限制性实施方案中,R1和R2可以是F。
附图简述
图1.互变异构化之后PLP-CPP-115加合物(右)和PLP-1加合物(左)以及主要附近残基的计算机模型(in silico model)。
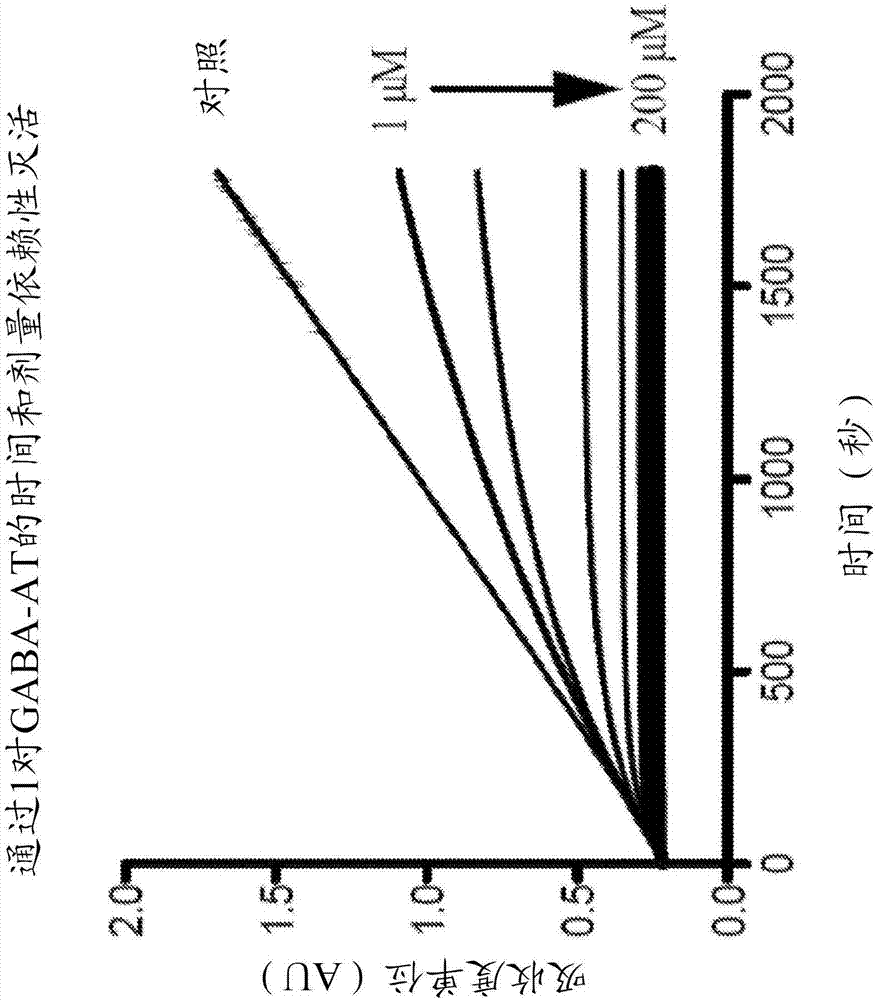
图2A-B.(A)通过1对GABA-AT的时间和浓度依赖性抑制。(B)k表观相对于浓度的二次标绘图(secondary plot)以确定1的k灭活值和KI值。
图3A-B.通过CPP-115在24h温育之后的灭活的GABA-AT的再活化(A)和通过1在4h温育之后的灭活的GABA-AT的再活化(B)。
图4.通过1对Asp-AT的浓度依赖性抑制。
图5.通过1对Ala-AT的浓度依赖性抑制。
图6A-B.(A)通过1对OAT的时间和浓度依赖性抑制。剩余的OAT活性的百分比的自然对数相对于预温育时间在每个抑制剂浓度被标绘以获得每个浓度的k表观(斜率)值。k表观是描述在每个抑制剂浓度的灭活的速率常数。(B)1的Michaelis-Mentel标绘图。使用非线性回归分析拟合k表观值以获得抑制常数(KI)和酶灭活的速率常数(k灭活)。
图7.通过1对hERG通道的抑制(hERG CHO-K1细胞系,检测方法:自动化膜片钳(automated patch-clamp)):0E-7M的测试浓度,上方;0E-6M的测试浓度,中间;0E-5M的测试浓度,下方。
图8.通过1对微粒体细胞色素P450的抑制。
图9A-B.人类肝微粒体(HLM)中的(A)特非那定和(B)1的时间依赖性损失。
图10A-C.对照(A);以及可卡因或尼古丁(B)和急性剂量的1和可卡因或尼古丁(C)对11C-雷氯必利摄取的效果的PET数字图像。
图11A-B.示出(A)可卡因和(B)可卡因和1对海马体中增加的代谢需求的效果的PET数字图像的统计学参数图。
某些实施方案的详述
当涉及本发明的某些实施方案时,用GABA-AT的计算机建模指示,未预料到地,如果环戊烷环构象通过环内双键(endocyclic double bond)的设置(installation)被锁住,那么CPP-115的二氟亚甲基基团将在互变异构化之后更接近Lys-329(用于1的相对于CPP-115的图1)。因此,本发明涉及(S)-3-氨基-4-(二氟亚甲基)环戊-1-烯-1-羧酸(1)及其结构变型、其用GABA-AT和非GABA能脱靶(non-GABAergic off-target)的生物学评估以及其在自由运动大鼠中关于在NAcc中的多巴胺的释放的体内活性,以及其对于神经元葡萄糖代谢的效果。
1的合成在方案2中被示出,该合成从CPP-115盐酸盐开始。羧酸基团和氨基基团首先被保护,并且然后甲酯的α-质子通过KHMDS来去质子化,随后添加苯基氯化硒(phenyl selenyl chloride),产生非对映异构体的7:3不可分离的混合物(4)。保护基团被除去以提供6。据发现,6的纯度对于1的最终纯度是重要的。在温和条件下6中的苯基硒基基团的氧化消除给出1和2的清洁的10:6异构体混合物。通过色谱法将1与2分离的许多尝试是不成功的,但是据发现在该过程中,2比1较不稳定。使用软的硫醇亲核体(2-巯基苯甲酸)选择性地改性并除去更有反应性的2的策略被成功地开发。在反应通过19F NMR被证实是完全的之后,使用C-18反相柱色谱法来提供纯的1。本发明和/或与本发明联合使用的多种其他化合物可以使用方案2中提供的种类的合成技术或其直接变型,从对应的3-氨基-4-亚甲基环戊烷-1-羧酸来制备,如将被本领域技术人员理解的并了解本发明。
方案2.从CPP-115合成1
初步体外结果示出1是GABA-AT的非常有效的灭活剂。因为灭活发生的如此迅速,所以通过1对GABA-AT灭活的抑制常数(KI)和酶灭活的速率常数(k灭活)不能使用常规的Kitz和Wilson再制图(Kitz and Wilson replot)来精确地确定,即使在非最优的条件下,如初始地对于CPP-115报告的。代替地,使用最近开发的进展曲线分析方法来测量动力学常数(图2),所述进展曲线分析方法允许在最优条件下的测量(Salminen,K.A.;J.;J.I.;Pasanen,M.;Auriola,S.;Juvonen,R.O.;Raunio,H.Drug Metab.Dispos.2011,39(3),412-418)。使用相同的方法来测量CPP-115的动力学常数作为参考。结果示出1具有比CPP-115更高的对GABA-AT的结合亲和力(1和CPP-115的KI值分别是9.7μM和59μM),并且1以比CPP-115更大的速率灭活GABA-AT(1和CPP-115的k灭活值分别是3.32min-1和2.05min-l)。总的来说,1的效率常数(efficiency constant)(k灭活/KI=342mM-1min-1)是CPP-115(k灭活/KI=34.9mM-1min-1)的9.8倍;因此,作为GABA-AT的灭活剂,1是CPP-115的效率的9.8倍。
因为1被设计成与Lys-329形成不可逆共价键,所以进行GABA-AT的时间依赖性再活化以测试该机制是否包括不可逆和/或可逆的抑制。在GABA-AT被10当量的1用4h温育完全灭活之后,灭活的酶被渗析并且在不同的时间间隔收集等分部分并测定酶活性的恢复。在72h的渗析之后,1灭活的GABA-AT的酶活性部分地恢复并稳定在20%(图3B)。预先对CPP-115进行GABA-AT的相同的时间依赖性再活化;当GABA-AT被100当量的CPP-115用24h温育完全灭活并然后渗析时,酶活性恢复至40%(图3A)。即使以10倍的浓度和长得多的温育时间,在灭活GABA-AT方面,CPP-115也比1低效得多。来自1灭活的GABA-AT的少量的酶活性的恢复指示,灭活可以包括不可逆组分(irreversible component)和可逆组分两者。
不同于氨己烯酸,CPP-115被报告不灭活或不抑制脱靶酶(off-target enzyme)例如天冬氨酸氨基转移酶(Asp-AT)和丙氨酸氨基转移酶(Ala-AT),这可能有助于其比氨己烯酸更大的安全系数。因此,1的活性在这些脱靶酶上被测试。结果示出1是Asp-AT和Ala-AT两者的非常弱的可逆抑制剂,其中IC50>4mM(图4和图5)。另一种重要的PLP-依赖性脱靶酶是鸟氨酸氨基转移酶(OAT);高水平的OAT损害通过尿素循环(urea cycle)由鸟氨酸氨甲酰基转移酶对氨的解毒。CPP-115被报告是温和的OAT的灭活剂,具有0.116mM的KI值和0.097min-1的k灭活值。化合物1还被示出是强效的OAT的灭活剂,具有0.0033mM的KI值和0.025min-1的k灭活值(图6)。通过比较1的k灭活/KI值(7.6mM-1min-1)与CPP-115的k灭活/KI值(0.84mM-1min-1),1是CPP-115的效率的9.0倍的OAT的灭活剂,这与其作为GABA-AT的灭活剂的较高效率一致。
hERG是有助于心脏的电活性的钾离子通道,心脏的电活性协调心跳。此通道对药物结合是敏感的,并且当该通道传导电流跨过细胞膜的能力被损害时,其可以导致可能致命的心脏副作用;因此,重要的是,在药物开发期间避免hERG抑制。与CPP-115相同,1不抑制hERG通道的活性(图7)。
微粒体细胞色素P450(CYP)是参与药物代谢的主要的酶,占全部药物代谢的~75%。因此,常进行微粒体稳定性以预测药物在药物开发期间是否将被太迅速地消除。与CPP-115相同,1不抑制或诱导参与药物代谢中的~95%的反应的七种最常见的CYP(1A、2B6、2C8、2C9、2C19、2D6以及3A)(图8)。血浆蛋白结合是仅27%,这指示血浆中高的游离药物百分比。
化合物1还被评估其在人类肝微粒体(HLM)中的代谢稳定性。这通过用微粒体温育1并使用LC-MS/MS监测其随着时间消失来完成。特非那定作为阳性对照以类似的条件运行。结果示出1是在HLM中稳定持续90min(图9)。
药物成瘾由当成瘾物质被摄取时NAcc中的多巴胺的释放造成。使用体内微正电子发射断层扫描(microPET)成像(micropositron emission tomography imaging)来确定1对自由运动大鼠中的多巴胺的释放的效果(图10)。(Dewey,S.L.;Morgan,A.E.;Ashby,C.R.;Horan,B.;Kushner,S.A.;Logan,J.;Volkow,N.D.;Fowler,J.S.;Gardner,E.L.;Brodie,J.D.Synapse 1998,30(2),119-129)。在其中存在高浓度的多巴胺D2受体(如通过图10中的每个图像的中间部分的两个灰色阴影斑点看出的)的中枢神经系统特别是纹状体中,[11C]-雷氯必利与多巴胺竞争位于突触后的多巴胺末端(post-synaptic dopamine terminal)的相同受体位点。使用microPET测量由可卡因或尼古丁诱导的突触多巴胺水平的升高引起的示踪剂[11C]-雷氯必利从多巴胺受体的解离(用可卡因和尼古丁获得相同的图像)。当动物接收可卡因(n=8)或尼古丁(n=6)时,纹状体多巴胺水平迅速升高。(Dewey,S.L.;Chaurasia,C.S.;Chen,C.E.;Volkow,N.D.;Clarkson,F.A.;Porter,S.P.;Straughter-Moore,R.M.;Alexoff,D.L.;Tedeschi,D.;Russo,N.B.;Fowler,J.S.;Brodie,J.D.Synapse 1997,25(4),393-398)。这些升高有效地从受体替代[11C]-雷氯必利,如在图10B中看出的(中间框;斑点是较少灰色阴影的(much less gray-shaded))。如果相同的动物(在不同日)在可卡因或尼古丁之前接收1,那么不存在[11C]-雷氯必利结合的变化(图10C,下框);斑点的阴影的程度(degree of shading)等于对照的程度(图10A,上框),这指示不存在将与[11C]-雷氯必利结合有效地竞争的多巴胺水平的升高。因此,1阻止可卡因和尼古丁两者诱导的多巴胺升高。
除了标记的雷氯必利研究之外,还使用[18F]-2'-氟-2'-脱氧-D-葡萄糖(18FDG)和microPET来检查1对于可卡因诱导的葡萄糖代谢的升高的区域性效果。18FDG是像葡萄糖一样被摄取到神经元(或人体内任何细胞)中的葡萄糖的类似物。然而,在18FDG被磷酸化之后,对应的6'-磷酸酯在糖酵解途径中不能被进一步代谢并保留在细胞中。因此,人类PET研究已经使用18FDG持续几十年来对大脑绘图(map)。例如,如果PET扫描器中的个体用一只手进行特定的任务,同时静脉注射18FDG,那么在作为那只手执行该任务的能力的基础的大脑中的神经元并入此放射标记的糖,而其他的周围的神经元没有并入。当人类或大鼠接收精神兴奋剂如可卡因时,多巴胺淹没突触,造成突触后神经元极度地兴奋(fire)。因为此神经元兴奋需要呈葡萄糖的形式的能量,所以结果是大脑葡萄糖代谢在特定的大脑区域增加。在自由运动大鼠中1对可卡因诱导的葡萄糖代谢的增加的效果使用统计学参数绘制来确定,其中来自仅可卡因的动物的所有图像被添加在一起并然后与从接收1和可卡因的相同的动物获得的图像相比。设定统计学阈值(p<0.00001),并产生统计学参数图,即示出在两个条件之间统计学上不同的所有像素的图像,并且然后覆盖在大鼠大脑的MRI上(图11)。在仅可卡因的动物中,观察到双侧型结构(bilateral structure)的海马体中的巨大活化,在每侧具有一个大的灰色斑点(图11A,左)。在可卡因/1动物中,海马体中的活化全部消失(图11B,右)。这是在此研究和有关的研究下观察到的通过化合物的摄取的最大衰减:氨己烯酸和CPP-115先前经历类似的测试;它们两者阻止可卡因诱导的纹状体多巴胺的增加,但不像1这样完全阻止海马体代谢。
已知可卡因和尼古丁诱导的纹状体多巴胺的增加产生条件性位置偏爱(CPP)(conditioned place preference),这导致动物‘学习’使特定的环境与它们接收的药物相关。当纹状体通过多巴胺水平升高来活化时(继可卡因或尼古丁挑战之后),向海马体的突出(projection)引起其活化。海马体在空间记忆方面起中枢作用;因此其对于在药物暴露期间编码环境条件是重要的。因为1阻止可卡因诱导的纹状体多巴胺的增加,因此不令人惊讶的是它还抑制海马体中的增加的代谢需求。
如可以与本发明的多个其他实施方案有关的,鸟氨酸氨基转移酶(OAT)属于与GABA-AT相同的PLP-依赖性酶的进化亚组(evolutionary subgroup)。这两种酶共享高的结构同源性,并且如同所有氨基转移酶,还具有非常类似的催化机制。如在共同未决的申请序号14/936,153中更充分地讨论的,OAT在许多组织中被表达,包括肝、肾、小肠、脑以及眼,并且催化鸟氨酸和α-酮戊二酸可逆转化成L-谷氨酸半醛,该L-谷氨酸半醛环化成Δ1-吡咯啉-5-羧酸盐和L-谷氨酸。然后,L-谷氨酸通过谷氨酰胺合成酶被转化成L-谷氨酰胺。
谷氨酰胺是体内最丰富的游离氨基酸;其对于正常细胞和赘生性细胞两者的生长是重要的。然而,肿瘤细胞比正常细胞更有效地摄取谷氨酰胺,并且肿瘤生长被谷氨酰胺增强。(参见例如,Souba,W.W.Glutamine and cancer.Ann.Surgery 1993,218,715-728;Medina,M.A.Glutamine and cancer.J.Nutr.2001,131(9增刊),2539S-2542S)。关于谷氨酰胺,癌症细胞将自身与正常细胞区分,因为它们对于谷氨酰胺具有增加的需求以支持刺激增殖的合成代谢过程。(2015年11月9日提交的前述'153申请通过引用以其整体并入本文)。
由于OAT和GABA-AT之间的结构相似性,已经示出某些GABA-AT的灭活剂还灭活OAT。如下文展示的,本发明的化合物也可以被用于调节、降低和/或抑制OAT活性。更具体地,在前述的并入的'153申请中详述的方法学和方案可以被用于示出如在治疗恶性病理性增殖紊乱中有用的此类化合物,所述恶性病理性增殖紊乱包括但不限于肝细胞癌。
如可以与本发明的多个其他实施方案有关的,GABA-AT抑制剂已经被示出对于治疗心理紊乱和神经紊乱及其组合是有效的,所述心理紊乱包括但不限于:广泛性焦虑紊乱(general anxiety disorder)、病理性或强迫性赌博紊乱(pathological or compulsive gambling disorder)、强迫性进食(compulsive eating)(肥胖症)、躯体变形障碍(body dysmorphic disorder)、疑病症(hypochondriasis)、病理性梳理状况(pathologic grooming condition)、盗窃癖(kleptomania)、纵火癖(pyromania)、注意力缺失多动症(attention deficit hyperactivity disorder)以及冲动控制障碍(impulse control disorder),所述神经紊乱包括但不限于癫痫、婴儿痉挛症、癫痫、部分发作、复杂性部分发作、继发性全身发作(secondary generalized seizure)、强直阵挛发作(tonic-clonic seizure)、琥珀酸半醛脱氢酶缺乏症(SSADHD)(succinic semialdehyde dehydrogenase deficiency)、韦斯特综合征中的婴儿痉挛症(infantile spasms in West's syndrome)、Lennox-Gastaut综合征、管状硬化(tubulous sclerosis)、图雷特综合征、运动紊乱(movement disorder)、纤维肌痛、神经病变、癫痫有关偏头痛、不宁腿综合征和创伤后应激障碍以及阿尔兹海默病,这样的治疗如在美国专利第8,969,413号中被描述,该专利的整体通过引用并入本文。因此,本发明的化合物还可以被用于治疗此类紊乱。更具体地,'413专利详述并并入到'413专利中的方法学和方案可以被用于示出如在治疗神经紊乱和心理紊乱中有用的此类化合物,所述神经紊乱和心理紊乱包括但不限于上文描述的那些。
本发明的方法还可以被扩展至或包括使用或联合药物组合物的方法,如将被本领域技术人员理解的,所述药物组合物包含本文描述的种类的化合物和生理学上或另外合适的制剂。在某些实施方案中,本发明包括连同在本文中被共同地称为载体的一种或更多种生理学上可耐受的或可接受的稀释剂、载体、佐剂或媒介物被配制成组合物的如上文陈述的一种或更多种GABA-AT或OAT灭活剂化合物。适合于这样的接触或施用的组合物可以包含生理学上可接受的无菌的水溶液或非水溶液、分散体、悬浮液或乳液。产生的组合物可以与本文描述的多种方法联合用于施用或接触细胞介质和/或其中表达的或以其他方式存在的GABA-AT或OAT。无论是否与药物组合物联合,“接触”意指为了将此类灭活剂化合物结合和/或络合至酶的目的,GABA-AT或OAT和一种或更多种灭活剂化合物被促使在一起。对此类氨基转移酶有效的化合物的量可以凭经验来确定,并且做出这样的确定在本领域技术内。调节、抑制或以其他方式影响GABA-AT或OAT活性包括降低和/或减轻两者以及消除GABA-AT活性和/或多巴胺释放,或可选择地降低和/或减轻两者以及消除OAT活性、谷氨酸产生、细胞增殖和/或肿瘤生长。
本领域技术人员将理解的是,剂量将随着特定灭活剂化合物的活性、疾病状态、施用途径、治疗持续时间以及在医学领域和药学领域熟知的类似因素而变化。通常,合适的剂量将是对产生治疗性或预防性效果有效的最低剂量的量。如果期望,此类化合物、其药学上可接受的盐或有关组合物的有效剂量可以以在合适的时间段内分开施用的两个或更多个亚剂量(sub-dose)来施用。
制备药物制剂或组合物的方法包括促使一种或更多种灭活剂化合物与载体和任选地一种或更多种另外的佐剂或成分联合的步骤。例如,可以使用标准药物配制技术,例如在Remington's Pharmaceutical Sciences,Mack Publishing Company,Easton,PA中描述的那些技术。
不考虑组合物或制剂,本领域技术人员将认识到用于药物施用的多种渠道,连同被认为致使此类药物适合于施用的对应的因素和参数。因此,关于一个或更多个非限制性实施方案,本发明提供一种或更多种灭活剂化合物用于制造用于在治疗多种疾病状态中的治疗性用途的药物的用途,特别地,关于GABA-AT,治疗神经紊乱和心理紊乱,包括成瘾和物质成瘾,和相关的指征,或关于OAT,治疗肝细胞癌或其预防。
发明的实施例
以下非限制性实施例和数据例证了与本发明的化合物/组合物和/或方法有关的各个方面和特征,包括多种GABA-AT和/或OAT灭活剂化合物,如通过本文描述的合成方法可获得的。与现有技术相比,本发明的化合物和方法提供令人惊讶的、未预料到的且与其相反的结果和数据。虽然本发明的实用性通过使用可以并入其中的若干化合物和取代基来例证,然而本领域技术人员将理解,采用如与本发明的范围是相称的多种其他化合物和取代基可获得相当的结果。
一般程序。CPP-115在慷慨地提供其的IRIX Pharmaceuticals for Catalyst Pharmaceuticals合成;其他化学品从Sigma-Aldrich获得并按原样使用,除非指明。所有合成使用火焰干燥的设备并采用在处理空气敏感性材料方面的标准技术、在无水条件下在氩气的气氛中进行,除非另外注明。将所有溶剂蒸馏并在使用之前储存在氩气或氮气气氛下。1H NMR和13C NMR光谱在Bruker AVANCE III 500光谱仪、Agilent DDR2 400MHz光谱仪或具有Agilent 5mm HFX探针的Agilent DD2 500MHz光谱仪上,在26℃使用DMSO-d6或D2O作为溶剂来获得,以δ(ppm)记录并参考DMSO-d6(对于1H NMR,2.50ppm,并且对于13C NMR,39.52ppm)或D2O(对于1H NMR,4.79ppm)。高分辨质谱(HRMS)用Agilent 6210 LC-TOF(ESI,APCI,APPI)质谱仪来测量。
实施例1
(1S,3S)-3-((叔丁氧基羰基)氨基)-4-(二氟亚甲基)环戊烷-1-羧酸甲酯(3)。在0℃,向干燥的甲醇(27mL)添加乙酰氯(2.49mL,35mmol)并搅拌持续10min。向产生的溶液添加CPP-115盐酸盐(1,1.5g,7.0mmol)并在室温搅拌持续24h。然后,添加三乙胺(6.8mL,49mmol)和二碳酸二叔丁酯(1.9mL,8.4mmol),并将产生的溶液在室温搅拌持续20h。将反应混合物浓缩并再溶解在乙酸乙酯中。有机溶液用2N HCl、饱和的NaHCO3和盐水洗涤。有机层经Na2SO4干燥,随后过滤并蒸发以提供作为白色固体的3(1.99g,6.83mmol,97%);1H NMR(500MHz,60℃,DMSO-d6)δ6.89(s,1H),4.57(s,1H),3.63(s,3H),2.86(m,1H),2.55–2.51(m,1H),2.23(ddt,J=10.0,7.1,2.9Hz,1H),1.79(m,1H),1.39(s,9H);13C NMR(126MHz,60℃,DMSO-d6)δ173.74,154.78,152.80,150.54,148.27,92.12,91.98,91.84,77.86,51.72,49.45,40.31,36.51,28.31,28.16;19F NMR(470MHz,60℃,DMSO-d6)δ-89.49(d,J=55.9Hz),-92.89(d,J=57.7Hz);对于C13H19F2NNaO4的HRMS(M+Na+)计算为314.1174,实测为314.1179。
实施例2
(3S)-3-((叔丁氧基羰基)氨基)-4-(二氟亚甲基)-1-(苯基硒基)环戊烷-1-羧酸甲酯(methyl(3S)-3-((tert-butoxycarbonyl)amino)-4-(difluoromethylenyl)-1-(phenylselanyl)cyclopentane-1-carboxylate)(4)。经由注射器向在-78℃的KHMDS(14.96mL的在THF中的1M溶液,14.96mmol)和干燥的THF(10mL)的溶液缓慢地添加3(1.98g,6.80mmol)在干燥的THF(10mL)中的溶液。将反应混合物在-78℃搅拌持续90min。添加苯基氯化硒(1.43g,7.48mmol)在干燥的THF(2mL)中的溶液,并且搅拌在-78℃继续持续75min。然后允许反应混合物加温至0℃,在0℃搅拌持续3h,加温至室温,并在室温搅拌持续2h。添加饱和的含水氯化铵和乙酸乙酯。有机层用饱和的含水氯化铵洗涤并经Na2SO4干燥。过滤和蒸发给出粗混合物,所述粗混合物通过硅胶柱色谱法(己烷/EtOAc)来纯化以提供作为浅棕色糖浆(syrup)的5:2非对映异构体混合物(4,2.12g,4.75mmol,70%);1H NMR(500MHz,60℃,DMSO-d6)δ7.58–7.37(m,5H),6.97(s,1H),4.83(s,0.7H),4.47(s,0.3H),3.64(s,2.2H),3.57(s,0.8H),2.96–2.88(m,1H),2.68–2.63(m,0.7H),2.43–2.36(m,0.7H),2.23–2.14(m,1.3H),2.00–1.95(m,0.3H),1.39(s,9H);13C NMR(126MHz,DMSO-d6)δ172.09,171.96,154.41,153.76,151.48,150.64,149.21,136.79,136.76,129.48,129.28,128.91,128.84,126.53,126.20,91.00,90.91,90.85,90.76,90.70,90.61,77.88,59.38,52.11,51.85,51.84,50.01,48.64,42.72,40.67,35.76,34.17,34.15,27.90,27.55;19F NMR(470MHz,60℃,DMSO-d6)δ-88.29(d,J=51.2Hz),-89.34(d,J=52.9Hz),-91.00(d,J=54.8Hz);对于C19H23F2NNaO4Se的HRMS(M+Na+)计算为470.0654,实测为470.0660(挑选最高丰度的Se同位素)。
实施例3
(3S)-3-((叔丁氧基羰基)氨基)-4-(二氟亚甲基)-1-(苯基硒基)环戊烷-1-羧酸(5)。在0℃向4(1.40g,3.13mmol)在甲醇(14mL)和水(4mL)中的溶液添加氢氧化锂(225mg,9.39mmol)。允许反应混合物加温至室温并搅拌持续20h。添加乙酸乙酯并且有机溶液用10%柠檬酸和盐水洗涤。然后将有机层经Na2SO4干燥,过滤并浓缩。使粗混合物经受硅胶柱色谱法(己烷/乙酸乙酯)以提供作为白色粉末的5(1.25g,2.89mmol,92%)。1H NMR(500MHz,60℃,DMSO-d6)δ12.61(s,1H),7.64–7.54(m,2H),7.46–7.42(m,1H),7.41–7.35(m,2H),6.94(s,1H),4.81(s,0.7H),4.48(s,0.3H),2.97–2.83(m,1H),2.67–2.56(m,0.7H),2.35(dd,J=16.9,2.7Hz,0.7H),2.20–2.10(m,1.3H),1.91(dd,J=12.9,8.3Hz,0.3H),1.42-1.35(m,9H);13C NMR(126MHz,DMSO-d6)δ173.76,173.56,154.71,154.64,153.93,151.65,150.69,149.38,136.87,136.81,129.60,129.39,129.20,129.14,127.06,126.55,91.67,91.52,91.40,91.37,91.26,91.11,78.06,52.38,49.99,48.69,48.42,42.95,40.68,36.08,34.26,28.15;19F NMR(470MHz,60℃,DMSO-d6)δ-88.52(d,J=52.9Hz),-89.53(d,J=53.2Hz),-91.25(d,J=53.2Hz),-91.45(d,J=55.3Hz);对于C18H21F2NNaO4Se的HRMS(M+Na+)计算为456.0502,实测为456.0498(挑选最高丰度的Se同位素)。
实施例4
3S)-3-氨基-4-(二氟亚甲基)-1-(苯基硒基)环戊烷-1-羧酸(6)。在0℃,向5(1.31g,3.03mmol)在CH2Cl2(13mL)中的溶液添加三氟乙酸(3.2mL)并将反应在相同的温度搅拌持续4h。将混合物浓缩并在真空干燥。使粗残余物经受阳离子交换柱色谱法(Dowex 50W-X8,5%含水吡啶作为洗脱剂)以提供作为灰白色固体的6(990mg,2.98mmol,98%)。1H NMR(500MHz,D2O)δ7.70–7.63(m,2H),7.53-7.46(m,1H),7.45-7.40(m,2H),4.61(t,J=6.7Hz,0.7H),4.44(m,0.3H),3.02–2.84(m,1.3H),2.71–2.57(m,1H),2.49(dd,J=14.7,7.8Hz,0.7H),2.29(dd,J=14.7,7.4Hz,0.7H),2.09(dd,J=14.4,5.3Hz,0.3H);13C NMR(126MHz,D2O)δ182.36,181.63,158.52,158.05,156.23,155.76,153.93,153.46,140.18,139.99,132.61,132.55,132.25,132.20,129.96,129.65,91.48,91.42,91.33,91.26,91.22,91.13,91.07,60.00,58.39,52.13,52.09,52.03,44.28,43.38,38.91,38.30;19F NMR(470MHz,D2O)δ-84.06(d,J=42.6Hz),-84.38–-84.58(m),-84.72(ddd,J=45.2,4.5,2.4Hz),-85.08(ddd,J=44.6,5.8,2.7Hz);对于C13H14F2NO2Se的HRMS(M+H+)计算为334.0158,实测为334.0155(选择最高丰度的Se同位素)。
实施例5
(S)-3-氨基-4-(二氟亚甲基)环戊-1-烯-1-羧酸盐酸盐(1)。在0℃向6(100mg,0.30mmol)和NaHCO3(55mg,0.66mmol)在水(2mL)中的溶液添加高碘酸钠(71mg,0.33mmol)。允许反应混合物加温至室温并搅拌持续6h。将反应混合物直接应用至阳离子交换柱(Dowex 50W-X8,2N HCl作为洗脱剂)以提供粗混合物。使粗混合物经受C-18反相柱色谱法(水/甲醇)以提供作为白色粉末的1和2的混合物(62mg,0.29mmol,96%)(注解:当浓缩样品时添加2N HCl的等分部分,以确保溶液为强酸性)。在0℃向1和2的混合物(51mg,0.24mmol)在甲醇(2mL)和水(0.5mL)中的溶液添加硫代水杨酸(112mg,0.72mmol)。允许反应混合物加温至室温并搅拌持续5h。在反应通过19F NMR证实为完全之后,将反应混合物浓缩并添加水。将悬浮液通过棉塞(cotton plug)过滤,并使滤液经受C-18反相柱色谱法(水/甲醇)以提供作为白色粉末的1(23mg,0.11mmol,从在先前异构体混合物中的1的含量的76%);1H NMR(500MHz,D2O)δ6.29(s,1H),5.16(s,1H),3.37(m,2H);13C NMR(126MHz,D2O)δ174.42,158.05,155.76,153.46,150.08,132.06,89.80,89.64,89.59,89.43,57.84,57.79,34.85;19F NMR(376MHz,D2O)δ-83.86(ddd,J=42.8,6.0,3.2Hz),-84.12(ddd,J=43.0 4.9,2.7Hz);对于C7H6F2NO2的HRMS(M-H-)计算为174.0372,实测为174.0374;HPLC纯度(100%,通过在210nm处的UV吸收度(UVabsorbance),100%,通过ELSD)。
实施例6
通过HPLC的样品纯度分析。使用Agilent 1260infinity HPLC系统,其由可变波长检测器(G1314A)、恒温柱室(thermostatted column compartment)(G1316A)、自动进样器(G1329B)、蒸发光散射检测器(ELSD,G4261A)、四元泵(quaternary pump)(G1311B)以及C-18反相柱(Agilent Poroshell 120,2.7μm,4.6mm×50mm)组成。实验用5μL(0.5mg/mL在水中)注射液进行,并且样品洗脱通过在210nm处的UV吸收度以及通过ELSD在线性梯度实验中来监测(具有0.05%三氟乙酸的水/乙腈,梯度体系:在7min内从初始的2%乙腈至100%乙腈,然后100%乙腈持续3min)。
实施例7
分子建模。所有绘制(rendering)在PyMol中进行。(Koo,Y.K.;Nandi,D.;Silverman,R.B.Arch.Biochem.Biophys.2000,374(2),248-254)。计算机模拟如先前描述的进行。(Silverman,R.B.;Bichler,K.A.;Leon,A.J.J.Am.Chem.Soc.1996,118(6),1241-1252)。简言之,配体使用R.E.D.服务器被制作(作为与辅因子的加合物)并使用AMBER程序的Antechamber模块被转化成拓扑文件(topology file)。(Yuan,H.;Silverman,R.B.Bioorganic Med.Chem.2006,14(5),1331-1338)。然后,使用Autodock 4.2将未互变异构化的分子对接(dock)到GABA-AT(由pdb条目#1OHW制备)的活性位点中,以Lys329作为柔性侧链。然后,使用GROMACS 4.5通过分子力学优化(refine)最好的对接结构。顺序(sequence)包括能量最小化、分子动力学(4ns)以及最终能量最小化。在此阶段,结构在合适的地方互变异构化,并且再次进行分子力学顺序。最终输出的结构被用于评估,而没有另外优化。
实施例8
酶和测定法。通过先前描述的程序,从猪脑纯化GABA-AT(1.48mg/mL)。(Koo,Y.K.;Nandi,D.;Silverman,R.B.Arch.Biochem.Biophys.2000,374(2),248-254)。使用已知的程序,从GABase(可商购的SSDH和GABA-AT的混合物)纯化琥珀酸半醛脱氢酶(SSDH)。(Silverman,R.B.;Bichler,K.A.;Leon,A.J.J.Am.Chem.Soc.1996,118(6),1241-1252)。GABA-AT活性使用公布的方法来测定。(Scott,E.M.;Jakoby,W.B.J.Biol.Chem.1959,234(4),932-936)。GABase(荧光假单胞菌(Pseudomonas fluorescens))和琥珀酸半醛购自Sigma-Aldrich。最终测定溶液由10mM GABA、1.2mM NADP+、5mMα-酮戊二酸、5mMβ-巯基乙醇以及在pH为8.5的50mM焦磷酸钾缓冲液中的过量SSDH组成。监测由NADP+转化成NADPH引起的在25℃在340nm处的UV吸收度的变化。使用1mm宽度、10mm路径长度、45mm高度微型石英比色皿,用Shimadzu UV-1800UV/Vis光谱仪记录用于确定k灭活值和KI值的酶测定。用BioTek Synergy H1酶标仪记录用于GABA-AT灭活和渗析实验的酶测定。
实施例9
k灭活值和KI值的确定。在对于1从1μM至200μM的范围内并且对于CPP-115从50μM至1600μM的范围内的不同浓度的灭活剂的存在下,在酶和测定法部分中描述的条件下测量GABA-AT的活性。使用GraphPad Prism 6TM软件,将由灭活造成的GABA-AT活性的曲线拟合成等式(1),以提供在每个灭活剂浓度的k表观值。
等式(1):
其中vi是初始速度,vS是稳态速度,t是时间,a0是初始吸收度并且k表观是观察到的灭活速率。(Salminen,K.A.;J.;J.I.;Pasanen,M.;Auriola,S.;Juvonen,R.O.;Raunio,H.Drug Metab.Dispos.2011,39(3),412-418)。将k表观值相对于化合物的浓度标绘,并且然后将最佳拟合曲线拟合成等式(2)以提供KI值和k灭活值。
等式(2):
其中[I]是灭活剂浓度,S是施加的底物(GABA)浓度,Km是底物(GABA)的Michaelis-Menten常数。用于计算的GABA与GABA-AT的Km值是1.3mM。(Yuan,H.;Silverman,R.B.Bioorganic Med.Chem.2006,14(5),1331-1338)。
实施例10
通过1对GABA-AT的灭活以及灭活的酶的渗析。渗析实验遵循先前报告的程序进行。(Lee,H.;Doud,E.H.;Wu,R.;Sanishvili,R.;Juncosa,J.I;Liu,D.;Kelleher,N.L.;Silverman,R.B.J.Am.Chem.Soc.2015,137(7),2628-2640)。向GABA-AT缓冲液(0.148mg/mL,30μL)添加50μL的16μM灭活剂缓冲溶液(pH 8.5的50mM焦磷酸钾、5mMα-酮戊二酸、5mMβ-巯基乙醇),使得GABA-AT和灭活剂的最终浓度将分别是1μM和10μM。在作为对照参考的另一个实验中,制备没有灭活剂的相同量的GABA-AT缓冲溶液。将样品溶液在黑暗中在室温温育持续4h。通过从该溶液取出5μL来测量剩余的酶活性。将灭活的GABA-AT溶液和对照GABA-AT溶液转移到D-TubeTM渗析器迷你型(Dialyzer Mini)(MWCO 12-14kDa)中,并在4℃相对于渗析缓冲液(350mL,50mM焦磷酸钾pH 8.5、0.1mMα-酮戊二酸、0.1mM 5'-磷酸吡多醛)进行渗析。渗析缓冲液在第4h、第8h和第24h交换三次。酶活性在第4h、第8h、第24h、第48h和第72h测量。
实施例11
通过1对天冬氨酸氨基转移酶的抑制。将微量滴定板孔加载有包含以pH 7.4的100mM磷酸钾、5.55mMα-酮戊二酸、1.11mM NADH、5.55mM L-天冬氨酸、11.1单位的苹果酸脱氢酶以及多种浓度的1的90μL的测定混合物。在室温温育混合物持续几分钟之后,添加10μL的Asp-AT(在pH 7.4的100mM磷酸钾中3.0单位/mL)。将板在室温摇动持续1min,并在340nm处每6s测量吸收度持续90min。所有测定一式两份地进行(图4)。
实施例12
通过1对丙氨酸氨基转移酶的抑制。测定与天冬氨酸氨基转移酶的相同,除了使用L-丙氨酸作为底物并且乳酸脱氢酶是酶(图5)。
实施例13
通过1对鸟氨酸氨基转移酶的时间和浓度依赖性的抑制。使用由Juncosa、Lee和Silverman的修改的程序来进行这些测定。用在总体积为20μL的包含1mMα-酮戊二酸的pH 8.0的100mM焦磷酸钾缓冲液中的多种浓度的1(0.5μM、2μM、5μM、10μM、20μM)在室温温育OAT(0.25μg)。在时间间隔处,将在pH 8.0的100mM焦磷酸钾缓冲液中的包含PYCR1(0.5μg)、12.5mMα-酮戊二酸、1mM NADH、0.03mM PLP和25mM L-鸟氨酸的、在37℃预温育持续10min的80μL的测定溶液添加至温育混合物并在37℃测定OAT活性持续20min。所有测定一式两份地进行,并且将在每个抑制剂浓度在每个预温育时间的剩余的OAT活性求平均。剩余的OAT活性的百分比的自然对数相对于预温育时间在每个抑制剂浓度被标绘以获得每个浓度的k表观(斜率)值。k表观是描述在每个抑制剂浓度的灭活的速率常数。使用非线性回归分析(GraphPad Prism 6TM;GraphPad Software Inc.)将k表观相对于抑制剂浓度再制图。KI和k灭活由等式(3)来评估:
等式3:
其中k灭活是最大灭活速率,KI是最大半灭活(half-maximal inactivation)所需的抑制剂浓度,并且[I]是1的预温育浓度(图6)。
实施例14
hERG通道的抑制。实验由Eurofins Panlabs(Redmond,WA 98052,USA)进行。使用hERG CHO-Kl细胞系。测试浓度是0.1μM、1μM和10μM。累积地,温育时间是在室温5min。检测方法使用自动化全细胞膜片钳。实验是一式两份的,并且将%尾电流(tail current)的抑制求平均(图7)。
实施例15
微粒体细胞色素P450的抑制。实验由Eurofins Panlabs(Redmond,WA 98052,USA)进行。测试CYP1A抑制(HLM,非那西汀底物)、CYP2B6抑制(HLM,安非他酮底物)、CYP2C8抑制(HLM,紫杉醇底物)、CYP2C9抑制(HLM,双氯芬酸底物)、CYP2C19抑制(HLM,奥美拉唑底物)、CYP2D6抑制(HLM,右美沙芬底物)以及CYP3A抑制(HLM,咪达唑仑和睾酮底物)。测试浓度是10μM。温育时间是在37℃的10min。检测方法是通过HPLC-MS/MS。实验是一式两份的,并且将%对照值的抑制求平均(图8)。
实施例16
microPET成像。成年雄性大鼠(Sprague-Dawley,200-250克,n=16)从Taconic Farms获得。将动物保持在12/12亮-暗循环中。使用Siemen's Inveon进行扫描。将所有发射扫描对于衰减进行校正。动物接收使用11C-雷氯必利或18FDG的基线microPET扫描,如先前描述的。(Patel,V.D.;Lee,D.E.;Alexoff,D.L.;Dewey,S.L.;Schiffer,W.K.Neuroimage 2008,41(3),1051-1066)。在动物是清醒的和自由运动时存在两种放射性示踪剂的吸收。在就要microPET扫描之前,将所有动物麻醉并保持在异氟烷下。
如展示的,本发明提供强效的GABA-AT灭活剂。特别地,体外结果示出1是CPP-115的9.8倍有效的GABA-AT的灭活剂,CPP-115是目前最有效的GABA-AT灭活剂,具有作为用于可卡因成瘾的治疗的高治疗性潜力。自由运动大鼠中的体内研究示出,在可卡因或尼古丁挑战之后,1在抑制NAcc中的多巴胺的释放方面优于CPP-115。化合物1还减弱多巴胺诱导的在海马体中代谢需求的增加,海马体是先前展示编码与药物诱导的多巴胺增加相关的环境的空间条件的脑部区域。
- 还没有人留言评论。精彩留言会获得点赞!