一种酮类化合物及其合成方法与流程
本发明属于有机合成技术领域,更具体地,涉及一种酮类化合物的方法。
背景技术:
邻二醇类化合物的碳碳键氧化断裂生成酮类化合物是有机合成的一类重要的反应(adv.carbohydr.chem.biochem.2006,60,183–250)。例如用biph3/nbs/k2co3,cro3,nis,ta(no3)3,can等作为氧化剂对邻二醇类化合物的碳碳键进行氧化断裂,反应温度为120℃(j.chem.soc.,chem.commun.,1981,1232–1233;can.j.chem.,1966,44,1323–1324;j.org.chem.,1981,46,1927–1929;j.org.chem.,1972,37,4204–4206;j.org.chem.,1969,34,869–871.)。基于以上相关国内外文献和专利的报道,所用的方法都需要高价化合物作为氧化剂,所用的氧化剂的毒性大,并且反应温度很高,底物适用性也不广泛,并不利于工业生产。
技术实现要素:
本发明的目的是为了克服现有技术的不足,提供一种邻二醇类化合物合成酮类化合物的方法。该方法只需在新型氧化剂的作用下,在一定的温度下搅拌反应,使邻二醇类化合物发生碳碳键断裂,即可得到酮类化合物。不需添加金属催化剂,反应温度低,没有副产物生成,具有高转化率,方便且环境友好型的特点。
本发明上述目的通过以下技术方案实现:
一种酮类化合物的合成方法,所述酮类化合物是邻二醇类化合物在溶剂的存在下,以1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐作为氧化剂,在70~90℃下搅拌,经萃取、干燥、过滤和减压蒸馏处理制得。
所述邻二醇类化合物的结构通式(1)为
优选地,所述r1为芳基或烷基,所述r2为卤基、烷基或硝基。
更为优选地,所述芳基为苯基或苯基上有甲基、硝基、氯、溴或氟取代,所述烷基为甲基,所述卤基为溴、氯或氟。
优选地,所述溶剂为n,n-二甲基甲酰胺、乙腈、四氢呋喃、1,4-二氧六环、1,2-二氯乙烷或甲苯。
优选地,所述邻二醇类化合物与溶剂的摩尔体积比为0.1:2mol/l;所述邻二醇类化合物和1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐的摩尔比为1:(2~3)。
优选地,所述搅拌的时间为20~24h,所述萃取的溶剂为乙酸乙酯,所述干燥的试剂为无水硫酸钠,所述干燥的温度为25℃,所述干燥的时间为10~15min。
所述减压蒸馏的温度为35~45℃,所述减压蒸馏的压力为-0.085~0.095mpa。
上所述方法合成的酮类化合物,所述酮类化合物的结构通式(2)为
优选地,所述r1为苯基或苯基上有甲基、硝基、氯、溴或氟取代,所述r2为甲基、溴、氯、氟或硝基。
与现有技术相比,本发明具有以下有益效果:
1.本发明采用1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐作为氧化剂使邻二醇类化合物的碳碳键发生氧化断裂,该氧化剂的毒性低,易于制备与保存,氧化效果明显,是其提供的氟正离子起到氧化的作用。
2.本发明采用氧化剂使邻二醇类化合物碳碳键断裂的方法合成酮类化合物,原料简单,反应可适底物范围广,合成简单,反应条件温和,产率高,副产物少,解决了现有技术中反应温度高,成本高,产物难以分离等问题,为邻二醇类化合物的碳碳键氧化断裂提供了新的高效方法。
附图说明
图1为实施例1,5,6,7中二苯甲酮的1hnmr图谱。
图2为实施例1,5,6,7中二苯甲酮的13cnmr图谱。
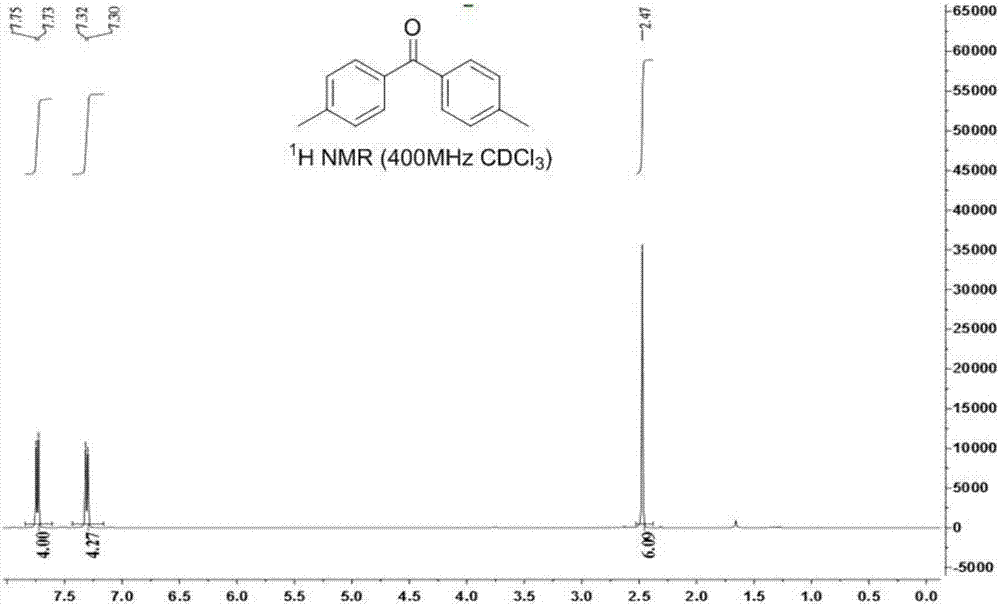
图3为实施例2中4,4’-二甲基二苯甲酮的1hnmr图谱。
图4为实施例2中4,4’-二甲基二苯甲酮的13cnmr图谱。
图5为实施例3中4,4’-二氯二苯甲酮的1hnmr图谱。
图6为实施例3中4,4’-二氯二苯甲酮的13cnmr图谱。
图7为实施例4中对硝基苯乙酮的1hnmr图谱。
图8为实施例4中对硝基苯乙酮的13cnmr图谱。
具体实施方式
下面通过实施例对本发明进行具体描述,有必要在此指出的是本实施例只用于对本发明进行进一步说明,但不能理解为对本发明保护范围的限制,该领域的技术熟练人员可以根据上述本发明的内容做出一些非本质的改进和调整。
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1合成二苯甲酮
称取0.1mmol四苯基乙二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐(selectflour)加入15ml耐压反应管中,加入磁力搅拌子和2mln,n-二甲基甲酰胺(dmf),80℃下搅拌反应20h,如式(1)所示。反应结束后用10ml水洗去溶剂,再用10ml乙酸乙酯萃取,用无水硫酸钠在25℃下干燥15分钟并过滤,最后用旋转蒸发仪在-0.09mpa,40℃下进行减压蒸馏除去有机溶剂,即可得到产物二苯甲酮,产率为90%。
图1为二苯甲酮的核磁表征图谱,其中,图1为二苯甲酮的1hnmr图谱,图2为二苯甲酮的13cnmr图谱。二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:7.47-7.51(t,4h),7.58-7.61(t,2h),7.80-7.82(d,4h)。13cnmr(100mhz,cdcl3)δ:128.4,130.2,132.6,137.7,196.9。
实施例2合成4,4’-二甲基二苯甲酮
称取0.1mmol1,1,2,2-四对甲苯基乙烷-1,2-二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2mln,n-二甲基甲酰胺,80℃下搅拌反应20h,如式(2)所示。反应结束后用10ml水洗去溶剂,再用10ml乙酸乙酯萃取,用无水硫酸钠在25℃下干燥15分钟并过滤,最后用旋转蒸发仪在-0.09mpa,40℃下进行减压蒸馏除去有机溶剂,即可得到产物4,4’-二甲基二苯甲酮,产率为85%。
4,4’-二甲基二苯甲酮的核磁表征图谱如图3和图4所示。其中,图3为4,4’-二甲基二苯甲酮的1hnmr图谱,图4为4,4’-二甲基二苯甲酮的13cnmr图谱。4,4’-二甲基二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:2.47(s,6h),7.30-7.32(d,4h),7.73-7.75(d,4h)。13cnmr(100mhz,cdcl3)δ:21.8,129.0,130.3,135.4,143.1,196.4。
实施例3合成4,4’-二氯二苯甲酮
称取0.1mmol1,1,2,2-四对氯苯基乙烷-1,2-二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2mln,n-二甲基甲酰胺,80℃下搅拌反应20h,如式(3)所示。反应结束后用10ml水洗去溶剂,再用10ml乙酸乙酯萃取,用无水硫酸钠在25℃下干燥10分钟并过滤,最后用旋转蒸发仪在-0.095mpa,35℃下进行减压蒸馏除去有机溶剂,即可得到产物4,4’-二氯二苯甲酮,产率为88%。
4,4’-二氯二苯甲酮的核磁表征图谱如图5和图6所示。其中,图5为4,4’-二氯二苯甲酮的1hnmr图谱,图6为4,4’-二氯二苯甲酮的13cnmr图谱。4,4’-二氯二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:7.46-7.48(d,4h),7.72-7.74(d,4h)。13cnmr(100mhz,cdcl3)δ:128.9,131.5,135.7,139.3,194.4。
实施例4合成对硝基苯乙酮
称取0.1mmol2,3-双(4-硝基苯基)丁烷-2,3-二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2mln,n-二甲基甲酰胺(dmf),80℃下搅拌反应20h,如式(4)所示。反应结束后用10ml水洗去溶剂,再用10ml乙酸乙酯萃取,用无水硫酸钠在25℃下干燥15分钟并过滤,最后用旋转蒸发仪在-0.085mpa,45℃下进行减压蒸馏除去有机溶剂,即可得到产物对硝基苯乙酮,产率为86%。
对硝基苯乙酮的核磁表征图谱如图7和图8所示。其中,图7为对硝基苯乙酮的1hnmr图谱,图8为对硝基苯乙酮的13cnmr图谱。对硝基苯乙酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:2.67(s,3h),8.09-8.11(d,2h),8.29-8.31(d,2h)。13cnmr(100mhz,cdcl3)δ:27.1,124.0,129.4,141.5,150.5,196.4。
实施例5合成二苯甲酮
称取0.1mmol四苯基乙二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2ml1,2-二氯乙烷(dec),90℃下搅拌反应22h,如式(5)所示。反应结束后用旋转蒸发仪在-0.09mpa,40℃下进行减压蒸馏除去有机溶剂,再用1ml二氯甲烷溶解残余物,最后通过硅胶柱层析即可得到产物二苯甲酮,产率为56%。
二苯甲酮的核磁表征图谱如图1图2所示,二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:7.47-7.51(t,4h),7.58-7.61(t,2h),7.80-7.82(d,4h)。13cnmr(100mhz,cdcl3)δ:128.4,130.2,132.6,137.7,196.9。
实施例6合成二苯甲酮
称取0.1mmol四苯基乙二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2ml乙腈(mecn),70℃下搅拌反应23h,如式(6)所示。反应结束后用旋转蒸发仪在-0.09mpa,40℃下进行减压蒸馏除去有机溶剂,再用1ml二氯甲烷溶解残余物,最后通过硅胶柱层析即可得到产物二苯甲酮,产率为75%。
二苯甲酮的核磁表征图谱如图1和图2所示,二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:7.47-7.51(t,4h),7.58-7.61(t,2h),7.80-7.82(d,4h)。13cnmr(100mhz,cdcl3)δ:128.4,130.2,132.6,137.7,196.9。
实施例7合成二苯甲酮
称取0.1mmol四苯基乙二醇,0.2mmol1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐加入15ml耐压反应管中,加入磁力搅拌子和2ml甲苯(toluene),80℃下搅拌反应24h,如式(7)所示。反应结束后用10ml水洗去溶剂,再用10ml乙酸乙酯萃取,用无水硫酸钠在25℃下干燥15分钟并过滤,最后用旋转蒸发仪在-0.09mpa,40℃下进行减压蒸馏除去有机溶剂,即可得到产物二苯甲酮,产率为82%。
二苯甲酮的核磁表征图谱如图1图2所示,二苯甲酮通过核磁表征与现有谱图特征结构得到确认。该化合物的表征数据如下:1hnmr(400mhz,cdcl3)δ:7.47-7.51(t,4h),7.58-7.61(t,2h),7.80-7.82(d,4h)。13cnmr(100mhz,cdcl3)δ:128.4,130.2,132.6,137.7,196.9。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合和简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!