一种三嵌段聚合物及其制备方法与流程
本发明涉及聚合物技术领域,尤其涉及一种三嵌段聚合物及其制备方法。
背景技术:
目前,肿瘤的治疗已成为亟待解决的全球性难题,在临床治疗中通常采取化疗手段进行治疗,对患者的身体状况有不同程度的不良影响,容易出现呕吐、掉发等不良反应,易产生耐药性,现已上市的白蛋白纳米粒等造价昂贵,肿瘤靶向性不理想,不能快速而有效的释放所负载的药物或基因,而且化疗手段易产生多药耐药、肿瘤无法切除干净、肿瘤的快速侵袭和转移等不良现象。聚合物胶束作为一种新型的药物载体,在医药等众多领域均显示出巨大的应用潜力,具有独特优势,其中比较突出的是较高的稳定性和较好的生物相容性,能增加难溶药物的溶解性,可作为基因的有效载体,降低毒副作用,具有被动和主动靶向作用,提高药物疗效。根据肿瘤微环境特点(如肿瘤细胞内外ph值、谷胱甘肽浓度、活性氧浓度、温度变化等),一些智能化聚合物胶束可在肿瘤细胞内发生刺激性响应,从而使结构破坏,释放所负载的化疗药物或基因,如genexol-pm已成功上市,另外也有多种聚合物胶束化疗制剂正处于临床研究中。
大部分化疗药物水溶性差,对肿瘤部位缺乏选择性,对正常组织或器官具有较强的毒副作用;如今,已出现使用sirna通过干扰细胞内基因的表达来逆转肿瘤的多药耐药、加速凋亡、极易转移等现象,但是sirna分子量较大,负电荷较强,极易被核酸酶降解等缺点,因此,选择合适的基因/药物载体十分重要。非病毒载体中由于阳离子聚合物其稳定性好,制备成熟,结构可调易控,且方便修饰等已成为基因药物或抗肿瘤药物的主要载体。报道的阳离子聚合物种类很多,如聚乙烯亚胺(pei)、聚酰胺、多聚赖氨酸、壳聚糖等。其中聚乙烯亚胺和聚酰胺表面有丰富氨基,可制备正电荷纳米粒子,与带负电荷的细胞膜相互作用,增加肿瘤细胞对载体的摄取,但该类聚合物通常具有较高的细胞毒性,一定程度上限制了其应用。
技术实现要素:
为解决上述技术问题,本发明的目的是提供一种具有智能响应功能的三嵌段聚合物及其制备方法。
本发明的三嵌段聚合物,结构如式(ⅰ)所示的聚合物,
其中,n=18-32,m=232-233。
本发明的三嵌段聚合物的制备方法,反应式如下:
其中,n=18-32,m=232-233。
包括以下步骤:
(1)壳寡糖(cso)与二硫代二丙酸(-ss-)在活化剂的作用下反应,得到第一中间体壳寡糖-二硫代二丙酸(cso-ss-)。
(2)步骤(1)中得到的第一中间体与聚乙烯亚胺(pei)在活化剂的作用下反应,得到第二中间体壳寡糖-二硫代二丙酸-聚乙烯亚胺(cso-ss-pei)。
(3)将步骤(2)中得到的第二中间体与咪唑丙烯酸(ua)在活化剂的作用下反应,得到式(ⅰ)的三嵌段聚合物壳寡糖-二硫代二丙酸-聚乙烯亚胺-咪唑丙烯酸(cso-ss-pei-ua)。
其中,步骤(1)中:
反应温度为30-45℃,优选40℃。
反应在溶剂中进行,溶剂为水、dmf、甲醇、氯仿和丙酮中的一种或几种,优选水和dmf。
反应时间为10-16h,优选12h。
cso与二硫代二丙酸的质量比1:2-3。
步骤(2)中:
反应温度为20-30℃,优选20℃。
反应在溶剂中进行,溶剂为水和磷酸盐缓冲液中的一种或两种,优选水,磷酸盐缓冲液优选ph7.4的磷酸盐缓冲液。
反应时间为10-16h,优选12h。
壳寡糖-二硫代二丙酸与pei的质量比为1:1.5-3,优选1:2。
步骤(3)中:
反应在溶剂中进行,反应温度为40-55℃,优选50℃。
溶剂为水、dmf、甲醇、氯仿和丙酮中的一种或几种,优选水和dmf。
反应时间为10-16h,优选12h。
壳寡糖-二硫代二丙酸-聚乙烯亚胺与咪唑丙烯酸的质量比为1.2-1.5:1,优选1.3-1.4:1。
步骤(4)中:
反应温度为20-30℃。
三嵌段聚合物的浓度为0.8-1.2g/ml,优选1mg/ml。
三嵌段聚合物的制备方法具体为:
(1)将壳寡糖(cso)溶于水中得到壳寡糖水溶液,并调节ph至7-8在搅拌条件下将其缓慢滴入活化剂和二硫代二丙酸(-ss-)dmf溶液的混合溶液中,并在30-45℃(优选40℃)下搅拌反应10-16h(优选12h),得到第一中间体壳寡糖-二硫代二丙酸(cso-ss-)。
其中,反应结束后,采用蒸馏水透析1-2天,优选24h(mwco=1000),透析液过滤,滤液冷冻干燥,产率约为95%。
所述cso分子量为3-5kda。
水优选蒸馏水。
ph可加微量的naoh调节。
(2)将步骤(1)中得到的第一中间体溶于水中得到第一中间体水溶液,并与活化剂、聚乙烯亚胺(pei)的水溶液混合,并在20-30℃下搅拌反应10-16h,优选12h,得到第二中间体壳寡糖-二硫代二丙酸-聚乙烯亚胺(cso-ss-pei)。
其中,反应结束后,采用透析袋(mwco=1000)透析1-2天,优选2天,冻干,产率约为90%。
pei水溶液加入第一中间体的水溶液中ph约为6时,约30min后再加入剩下的pei水溶液。
pei分子量为600g/mol。
水优选蒸馏水。
(3)将步骤(2)中得到的第二中间体溶于水中得到第二中间体水溶液,并缓慢滴加到活化剂和咪唑丙烯酸(ua)dmf溶液的混合溶液中,在40-55-℃(优选50℃)下搅拌反应10-16-h,优选12h,得到式(ⅰ)的三嵌段聚合物壳寡糖-二硫代二丙酸-聚乙烯亚胺-咪唑丙烯酸(cso-ss-pei-ua)。
其中,反应结束后,将液体置于透析袋中(mw=1000),蒸馏水透析2-3d,优选2d,透析液过滤,取上清液冷冻干燥,产率约为90%。
ua分子量为138.12g/mol。
水优选蒸馏水。
进一步的,步骤(1)中,所述二硫代二丙酸经活化剂活化羧基后与所述壳寡糖发生反应。
步骤(2)中,所述第一中间体经活化剂活化羧基后与聚乙烯亚胺发生反应。
步骤(3)中,所述咪唑丙烯酸经活化剂活化羧基后与所述第二中间体发生反应。
进一步的,所述活化剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edc)盐酸盐、n-羟基丁二酰亚胺(nhs)、二环己基碳二亚胺(dcc)中的一种或几种。
优选edc和nhs,两者的摩尔比为1:1。
进一步的,步骤(1)中,活化剂与-ss-的摩尔比为0.8-1.2:1,优选1:1,二硫代二丙酸中的羧基摩尔量过量,可保证只反应一个羧基。
步骤(1)中,在30-45℃下,优选40℃,所述二硫代二丙酸溶液在活化剂中进行羧基活化约30min。
进一步的,步骤(2)中,活化剂与第一中间体cso-ss-的摩尔比为0.8-1.2:1,优选1:1。
步骤(2)中,将第一中间体cso-ss-水溶液与活化剂水溶液混合,搅拌反应进行羧基活化。
进一步的,步骤(3)中,活化剂与咪唑丙烯酸的摩尔比0.8-1.2:1,优选1:1。
步骤(3)中,在40-55℃下,所述咪唑丙烯酸溶液在活化剂中进行羧基活化约30min。
进一步的,步骤(1)中,反应前,所述壳寡糖的浓度为20-40mg/ml,优选25-35mg/ml,进一步优选30mg/ml。
进一步的,步骤(1)中,反应前,所述-二硫代二丙酸-的浓度为65-80mg/ml,优选70-80mg/ml,进一步优选70mg/ml。
进一步的,步骤(2)中,反应前,所述壳寡糖-二硫代二丙酸浓度为5-15mg/ml,优选8-12mg/ml,进一步优选0mg/ml。
进一步的,步骤(2)中,反应前,所述聚乙烯亚胺是浓度为350-450mg/ml,优选400-450mg/ml,进一步优选400mg/ml。
进一步的,步骤(3)中,反应前,所述壳寡糖-二硫代二丙酸-聚乙烯亚胺浓度为4-6mg/ml,优选4-5mg/ml,进一步优选4.8mg/ml。
进一步的,步骤(3)中,反应前,所述咪唑丙烯酸浓度为15-20mg/ml,优选17-18mg/ml,进一步优选17.2mg/ml。
借由上述方案,本发明至少具有以下优点:
本发明所采用的cso具有良好的生物相容性和水溶性,可作亲水性外壳,但单独使用作为载体极不稳定;pei广泛的应用于基因载体,具有质子海绵效应,但单独使用毒性较大,因此为了解决这一问题,本发明将二硫代二丙酸中的两个羧基活化,作为连接臂分别链接cso和pei,相对于pei,cso的接枝率为3.33%,ua的接枝率为44.4%,pei一端连接咪唑丙烯酸(ua)为疏水内核可用来包载难溶性药物,另一端通过二硫代二丙酸中的二硫键(-ss-)外包一层cso用来维持整个聚合物为正电,同时降低pei的毒性,聚合物可在水中自组装形成具有ph敏感和还原敏感的纳米粒子,从而降低了聚合物的毒性和体现了聚合物在肿瘤微环境中的还原敏感性(-ss-)和ph敏感性(质子海绵效应),采用pei为主链可用来吸附sirna(各单元之间均是通过edc和nhs活化羧基,使氨基与羧基反应形成酰胺键),以此构成的载体可实现肿瘤的药物和基因联合治疗,为肿瘤的递药系统提供一种新型载体。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
图1是本发明的实施例1中cso(a)、cso-ss-(b)、pei(c)、cso-ss-pei(d)、cso-ss-pei-ua(e)的1h-nmr图谱;
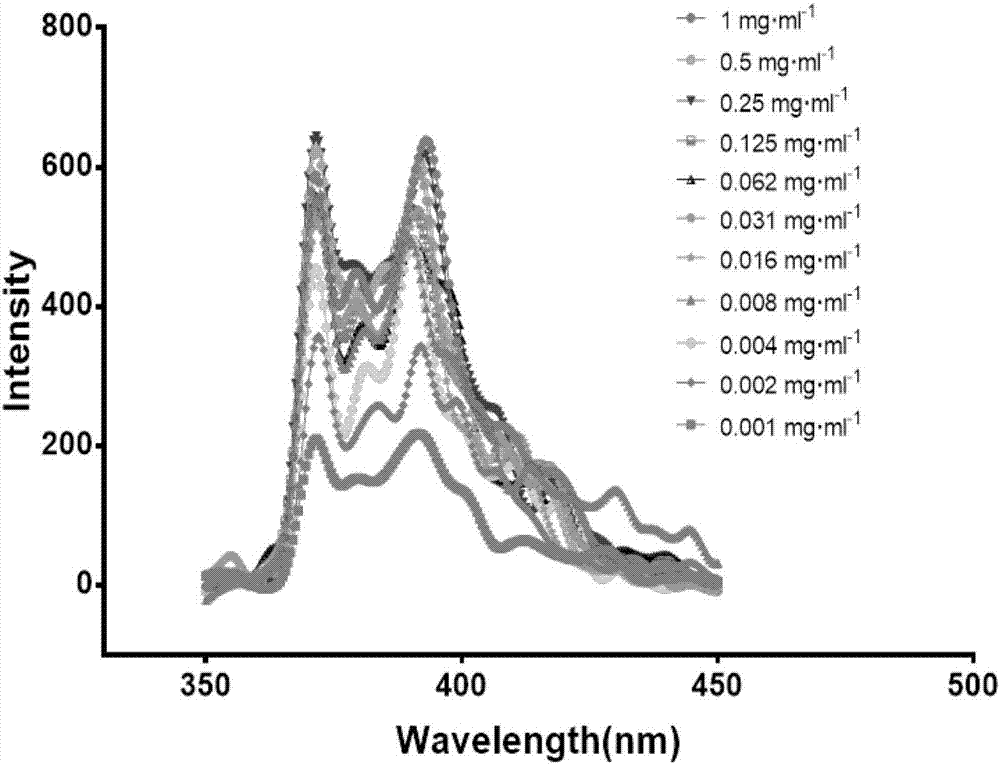
图2图示了本发明的实施例1中三嵌段聚合物在不同浓度时的荧光强度;
图3图示了本发明的实施例1中三嵌段聚合物在λ1=372nm处和λ3=383nm处的荧光强度的商(i1/i3)与相应浓度的对数的比值;
图4图示了本发明的实施例1中三嵌段聚合物在动态光散射粒度分析仪dls检测的粒径大小变化。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1
三嵌段聚合物的制备,包括以下步骤:
(1)cso-ss-的合成
将1.578g二硫代二丙酸、1.437gedc和0.861gnhs溶于20mldmf中,40℃活化30min待用;称取0.6gcso分散于20ml蒸馏水中,加入微量naoh调其ph为7-8,搅拌条件下将其缓慢滴入二硫代二丙酸溶液中,40℃搅拌反应12h后,用蒸馏水透析1d(mwco=1000),透析液过滤,滤液冷冻干燥,即得cso-ss-,产率为95%。
(2)cso-ss-pei的合成
将1g步骤(1)制备的cso-ss-溶解于100ml蒸馏水中,搅拌形成水溶液;将2.935gedc、1.435gnhs溶于5ml蒸馏水中形成活化剂水溶液,并将其加入cso-ss-水溶液中活化30min,将2gpei溶于5ml蒸馏水中,将其加入活化后的cso-ss-水溶液中,并当ph到6左右时停止加入,30min后再加完剩下的pei水溶液,20℃搅拌反应12h,1000透析袋透析(mwco=1000)2天,透析液过滤,滤液冷冻干燥,即得cso-ss-pei,产率为90%。
(3)cso-ss-pei-ua的合成
将0.43gua、1.19gedc和0.71gnhs加入到含有25mldmf的烧瓶中,搅拌30min至其溶解;将0.6g步骤(2)中制备的cso-ss-pei-ua溶解于125ml蒸馏水中,然后将其缓慢滴加到上述烧瓶中,50℃下反应12h,反应结束后将烧瓶内的液体置于透析袋中(mw=1000),蒸馏水透析2d,透析液过滤,取上清液冷冻干燥,得cso-ss-pei-ua,产率为90%。
下面对该实施例中的化合物进行表征以及产物的性能测定:
1、核磁表征
将cso、cso-ss-、pei、cso-ss-pei和cso-ss-pei-ua分别溶于d2o中,在400mhz核磁共振仪上测试,1h-nmr图谱如图1所示:cso中δ=4.67(h1)3.20-4.00ppm(糖环),2.01(-ch2);δ=2.74,2.93ppm处的信号归属于二硫代二丙酸中的五杂环基团的特定质子峰,表明二硫代二丙酸成功接枝到cso;cso-ss-pei共轭谱图中δ=2.3-2.7ppm处的质子峰证实了pei的引入;ua的信号位于δ=8.23(ha),δ=7.61(hb),δ=7.5(hc)和δ=6.51(h1),表明ua成功引入cso-ss-pei;给定cso-ss-pei-ua的光谱,分配给上述所有结果的峰意味着cso-ss-pei-ua共聚物的成功合成,cso-ss-pei-ua聚合物已成功制备。
2、聚合物的临界胶束浓度(cmc)测定
以芘为疏水性荧光探针测定聚合物的在ph7.4时的cmc值,以测定的各样品溶液的浓度对数值(lgc)为横坐标,如图2所示,聚合物在不同浓度时的荧光强度,以各样品溶液在λ1=372nm处和λ3=383nm处的荧光强度的比值(i1/i3)为纵坐标,绘制散点图,如图3所示,根据各点作出数据点的水平切线,以及突变曲线的切线,两个切线的交点所对应的聚合物浓度,即为临界胶束浓度(cmc)。
从图3可知,当聚合物浓度较低时,i1/i3值保持不变,表明聚合物未形成胶束;当浓度达到一定值以后,i1/i3值急剧下降,说明聚合物在该浓度时开始形成胶束。与小分子表面活性剂相比,聚合物的cmc可以达到7.94×10-3mg.ml-1,这说明在稀释过程中,该聚合物形成的胶束相对稳定,有作为药物载体的可能性。
3、聚合物的ph敏感性和还原敏感性测定
通过探针超声处理方法制备聚合物纳米粒子(2mg/ml),为了研究聚合物纳米粒子的ph和氧化还原响应行为,我们用动态光散射粒度分析仪dls检测粒径的大小变化,如图4所示。将所测试的聚合物纳米粒子在下列条件下溶解在不同的溶液中:(i)在ph7.4下,(ii)在ph5.3下,(iii)在ph7.4下用谷胱甘肽(gsh)(100mm)和(iv)在ph5.3下gsh(100mm)。
聚合物纳米粒子分别与磷酸盐缓冲液(pbs)(ph7.4)+gsh和pbs(ph5.3)孵育1小时后,从124.6nm膨胀至365.3和622.6nm,对于pbs(ph5.3)+gsh获得双峰,这暗示了胶束稳定性被破坏。粒径的变化是由于咪唑基在酸性溶液中的质子化,并且在gsh的存在下发生二硫键的断裂,这导致亲水/疏水平衡的快速分解并促进药物和基因的快速细胞内释放。
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!