碳硅烷聚合物的制作方法
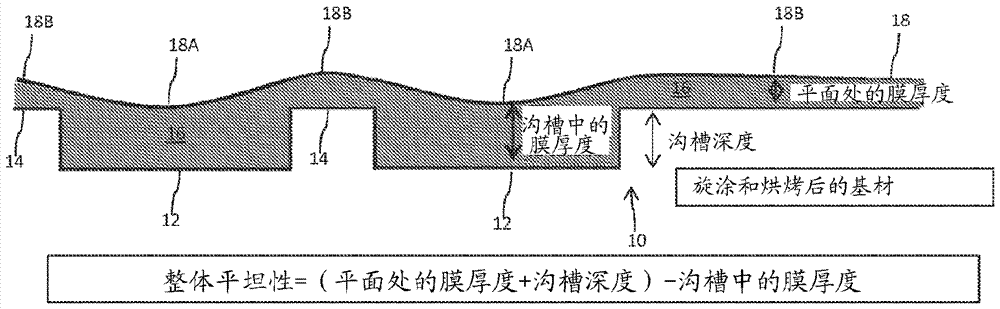
相关申请的交叉引用本申请根据标题35,u.s.c.§119(e)要求于2014年12月1日提交的标题为碳硅烷聚合物的美国临时申请序列62/085,892的权益,该申请的全部公开内容通过引用的方式明确地并入本文。发明领域本公开内容大体上涉及碳硅烷聚合物,更具体地涉及由碳硅烷单体组分和贡献羰基的单体形成的碳硅烷聚合物。背景在先进的半导体制造方法中,存在对高度平坦化材料的日益增长的需求,所述材料不仅提供窄间距形貌的无空隙填充,而且还能够提供平坦的表面。这些材料可以是具有反射控制特性的底部抗反射涂层(barc)。此外,所述材料可以是牺牲性的,其中它必须可通过湿去除化学物质选择性除去而不损坏下层或其他暴露的膜或基材。图1a图示说明了一个待用平坦化涂层涂覆的示例性基材10。图1a还示出了多个通过基材10的表面上的要素(feature)14隔开的说明性沟槽12。在施加和烘烤之后的施加的涂层16的一个理想情况示于图1b中。在该理想情况下,无论表面18a位于沟槽12的上方还是表面18b位于要素14的上方,涂层16的表面18都具有完全平坦的涂层。这样的理想情况不可能实现。在施加和烘烤之后的施加的涂层16的一个更典型的情况示于图1c中。在该典型的情况下,涂层16的表面18不是完全平坦的,并且至少部分地符合沟槽12和要素14的高度。例如,位于沟槽12上方的表面18a通常低于位于要素14上方的表面18b。可以由下式计算施加的涂层16的整体平坦性值:整体平坦性=(在要素的中心处测量的最宽的要素的顶部上的膜厚度+沟槽深度)-在最宽的沟槽的中心处的膜厚度。当整体平坦性值接近零时,涂层16的表面18接近完全平坦的涂层,如在图1b中图示说明的。通常,较低的整体平坦性值是优选的。接下来参考图2a,其图示说明了更加复杂的包括沟槽12和要素14的基材20。基材20示例性地包括第一区域和第二区域24,第一区域包含一个或多个相对窄的沟槽12a,第二区域包含一个或多个相对宽的沟槽12b。在施加和烘烤之后的典型的施加的涂层16示于图2b中。如图2b中图示说明的,涂层16的表面18不是完全平坦的,但是第一区域22上方的表面18比第二区域24上方的表面18更加平坦。图2b中的表面18的平坦性可以由下式计算:最宽的要素的顶部上的中心处的膜厚度-最窄的要素的顶部上的中心处的膜厚度。上式相当于图2b中的(a-b)。或者,图2b中的表面18的平坦性可以由下式计算:(邻近宽要素的空间的顶部上的膜厚度+宽要素的高度)-宽要素的中心的膜厚度。上式相当于图2b中的(b+c)-d。前述改善是期望的。发明概述本公开内容提供包含由至少一种碳硅烷单体组分和至少一种贡献羰基的单体形成的碳硅烷聚合物的组合物。在一些实施方案中,所述组合物适合作为间隙填充和平坦化材料,并且可任选地包含至少一种发色体(chromophore)用于照相平版应用。在一个示例性实施方案中,通过在适当的反应介质中将一种或多种单体组合导致形成均聚物或共聚物来形成有机碳硅氧烷膜的牺牲性旋涂层(sacrificialspin)。在安全且常见的工业溶剂的溶剂共混物中将一种或多种烷氧基单体组合,将酸溶液加入所述溶剂共混物中以催化水解-缩合反应。在优化的时间和温度下加热该反应溶液以形成低分子量且稳定的聚合物。在一个示例性实施方案中,通过并入一种或多种吸收248nm或193nm波长的uv光的发色体形成吸收248nm或193nmuv的配制物。在一些实施方案中,该配制物具有约800-约2500amu的分子量范围。在一些实施方案中,该分子量范围提供期望的高的湿蚀刻速率和等离子体蚀刻速率。根据本公开内容的一个实施方案,组合物包含碳硅烷聚合物,其中所述碳硅烷聚合物由至少一种碳硅烷单体和至少一种贡献羰基的单体形成。在一个实施方案中,所述碳硅烷聚合物具有10wt.%-45wt.%的硅含量或3wt.%或更大的羰基含量,基于聚合物的总重量。在一个更具体实施方案中,所述碳硅烷聚合物具有10wt.%-45wt.%的硅含量。在一个更具体的实施方案中,所述碳硅烷聚合物具有3wt.%或更大的羰基含量。在一个更具体的实施方案中,所述碳硅烷聚合物具有10wt.%-45wt.%的硅含量以及3wt.%或更大的羰基含量。在任一个上述实施方案的一个更具体的实施方案中,所述碳硅烷聚合物具有13wt.%-30wt.%的硅含量以及3wt.%或更大的羰基含量。在任一个上述实施方案的一个更具体的实施方案中,所述碳硅烷单体具有式:其中:x选自直链或支链的c1-c12烷基或c6-c14芳基,并且每个r是可水解的基团(反应性基团并通过该基团产生交联)或不参与交联的末端端基。在又一更具体的实施方案中,所述碳硅烷单体是双(三乙氧基甲硅烷基)乙烷。在任一个上述实施方案的一个更具体的实施方案中,所述贡献羰基的单体选自丙烯酸类单体、含羧基的单体和酸酐单体。在一个更具体的实施方案中,所述贡献羰基的单体是甲基丙烯酰氧基丙基三甲氧基硅烷。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种交联促进剂。在一个甚至更具体的实施方案中,所述交联促进剂是具有式:si(or)3(ch2)nnh3+(f3cso3)-的氨基硅烷盐,其中n是1-10的整数,每个r独立地为c1-c20烷基。在一个更具体的实施方案中,所述交联促进剂是氨基丙基三乙基硅烷。在又一更具体的实施方案中,所述交联促进剂是apteos三氟甲磺酸盐。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种溶剂。在一个甚至更具体的实施方案中,所述溶剂包含平坦化增强剂,例如碳酸烷基酯。在又一更具体的实施方案中,所述平坦化增强剂包括丙二醇碳酸酯。在任一个上述实施方案的一个更具体的实施方案中,所述碳硅烷聚合物具有1,000或更小的分子量。在任一个上述实施方案的另一更具体的实施方案中,所述碳硅烷聚合物具有约800-约1500、约800-约2500或约800-约5000的分子量。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种发色体。在一个更具体的实施方案中,所述发色体包括pteos和tesac中的至少一种。在另一实施方案中,所述组合物不包含发色体。在任一个上述实施方案的一个更具体的实施方案中,所述碳硅烷聚合物进一步由至少一种有机烷氧基硅烷单体形成。在一个甚至更具体的实施方案中,所述有机烷氧基硅烷单体选自甲基三甲氧基硅烷(mtmos)、甲基三乙氧基硅烷(mteos)、二甲基二乙氧基硅烷(dmdeos)、苯基三乙氧基硅烷(pteos)、二甲基二甲氧基硅烷、苯基三甲氧基硅烷、二苯基二乙氧基硅烷、二苯基二甲氧基硅烷和9-蒽羧基-烷基三烷氧基硅烷。根据本公开内容的另一实施方案,通过将任一个上述实施方案施加至表面上并且烘烤所述组合物以形成膜而形成膜。根据本公开内容的另一实施方案,提供了形成组合物的方法。所述方法包括使至少一种碳硅烷单体和至少一种贡献羰基的单体反应以形成碳硅烷聚合物。在一个更具体的实施方案中,所述碳硅烷聚合物具有10wt.%-45wt.%的硅含量。在另一更具体的实施方案中,所述碳硅烷聚合物具有3wt.%或更大的羰基含量。在又另一更具体的实施方案中,所述碳硅烷聚合物具有13wt.%-30wt.%的硅含量和3wt.%或更大的羰基含量。在一个更具体的实施方案中,所述方法包括使所述单体在约50℃-90℃的温度下反应约1小时-约5小时。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种溶剂。在一个甚至更具体的实施方案中,所述溶剂包含平坦化增强剂,例如碳酸烷基酯。在又一更具体的实施方案中,所述平坦化增强剂是丙二醇碳酸酯。在一个示例性实施方案中,提供了组合物。所述组合物包含至少一种选自碳硅烷单体、贡献羰基的单体和有机烷氧基硅烷单体的单体;和至少一种溶剂,其中所述溶剂包含平坦化增强剂,例如碳酸烷基酯。在一个更具体的实施方案中,所述平坦化增强剂包括丙二醇碳酸酯。在一个更具体的实施方案中,所述溶剂包含第一溶剂诸如pgmea或异戊醇,和丙二醇碳酸酯。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含发色体。在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含硝酸。在任一个上述实施方案的一个更具体的实施方案中,所述溶剂包含第一溶剂和平坦化增强剂诸如丙二醇碳酸酯。在任一个上述实施方案的一个更具体的实施方案中,至少一种单体包括至少一种选自甲基三甲氧基硅烷(mtmos)、甲基三乙氧基硅烷(mteos)、二甲基二乙氧基硅烷(dmdeos)、苯基三乙氧基硅烷(pteos)、二甲基二甲氧基硅烷、苯基三甲氧基硅烷、二苯基二乙氧基硅烷、二苯基二甲氧基硅烷和9-蒽羧基-烷基三烷氧基硅烷的有机烷氧基硅烷单体。在任一个上述实施方案的一个更具体的实施方案中,至少一种单体包括至少一种选自btse、1,2-双(三乙氧基甲硅烷基)甲烷、4,4-双(三乙氧基甲硅烷基)-1,1-联苯和1-4-双(三乙氧基甲硅烷基)苯的碳硅烷单体。在任一个上述实施方案的一个更具体的实施方案中,至少一种单体包括至少一种选自丙烯酸类单体、含羧基的单体或含酸酐的单体的贡献羰基的单体。在一个甚至更具体的实施方案中,所述至少一种单体包括甲基丙烯酰氧基丙基三甲氧基硅烷。附图简述通过结合附图参考本发明的实施方案的以下描述,本公开内容的上述和其他特征和优点以及获得它们的方式将变得更加明显,并且将更好地理解本发明,其中:图1a图示说明了在涂覆之前的示例性基材。图1b图示说明了施加于图1a的示例性基材的理想涂层。图1c图示说明了施加于图1a的示例性基材的另一涂层。图2a图示说明了包含低密度区域和高密度区域的另一示例性基材。图2b图示说明了施加于图2a的示例性基材的涂层。贯穿于若干视图,相应的附图标记指示相应的部分。本文所阐述的范例举例说明本发明的示例性实施方案并且这样的范例不应被解释为以任何方式限制本发明的范围。详细说明a.间隙填充和平坦化材料在一个示例性实施方案中,间隙填充或平坦化材料由组合物形成。所述组合物包含碳硅烷聚合物。所述组合物可以任选地包含交联促进剂、溶剂、发色体或催化剂中的一种或多种。在一些示例性实施方案中,所述材料作为间隙填充或平坦化层形成于适当的基材上。示例性的基材包括介电膜、多晶硅膜、介电-金属层、金属-硅层或有机层,例如位于在半导体制造方法中使用的硅晶片上。在一些示例性实施方案中,形成的层具有约61、约58、约48、或更小或在由前述值中的任意两个限定的任意范围内的平坦性值。在一个示例性实施方案中,形成的层具有大至约500nm、约400nm、约300nm、小至约200nm、约100nm、约70nm或在由前述值中的任意两个限定的任意范围内的厚度。在一个示例性实施方案中,形成的层在含水的碱性剥离剂化学物质中是牺牲性的,所述化学物质例如为在升高的温度下的氢氧化铵或者j.t.bakerclk-888stripperandresidueremover,可自avantorperformancematerials获得,但是耐室温下的2.3含水的四甲基氢氧化铵(tmah)、乙酸正丁酯(nba)、在40℃和70℃下的sc1(按照1/18/60的体积比的29%氢氧化铵+31%过氧化氢+di水)和丙二醇甲醚乙酸酯(pgmea)。b.碳硅烷聚合物在一个示例性实施方案中,所述间隙填充或平坦化材料由包含碳硅烷聚合物的组合物形成。所述碳硅烷聚合物包含碳硅烷单体和贡献羰基的单体。在一个实施方案中,按湿重计,所述碳硅烷聚合物占所述聚合物的总重量的少至约0wt.%、约1wt.%、约15wt.%、约30wt.%、多至约80wt.%、约90wt.%、约99wt.%、约100wt.%、或在由前述值中的任意两个限定的任意范围内,例如1wt.%-99wt.%,15wt.%-90wt.%,或30wt.%-80wt.%。在一个示例性实施方案中,所述碳硅烷聚合物是所述碳硅烷单体和贡献羰基的单体单元的包含具有变化尺寸的低聚物单元的无规共聚物。在另一示例性实施方案中,所述碳硅烷聚合物是具有规则的交替碳硅烷单体和贡献羰基的单体单元的交替共聚物。在又另一示例性实施方案中,所述碳硅烷聚合物是包含硅烷单体和贡献羰基的单体单元的嵌段共聚物。在一个示例性实施方案中,所述碳硅烷聚合物具有基于聚合物的总重量的少至约10wt.%、约13wt.%、约15wt.%、约20wt.%、多至约25wt.%、约30wt.%、约45wt.%、或在由前述值中的任意两个限定的任意范围内的硅含量,例如约10wt.%-约45wt.%,或约13wt.%-约30wt.%。在一个示例性实施方案中,所述碳硅烷聚合物具有约3wt.%、约5wt.%、约10wt.%、约13wt.%、约14wt.%、约15wt.%、约20wt.%或更大、或在由前述值中的任意两个限定的任意范围内的羰基含量,例如约3wt.%-20wt.%,约5wt.%-约15wt.%,约10wt.%-约15wt.%,或约13wt.%-约14wt.%。在一个实施方案中,所述碳硅烷聚合物具有少至约10wt.%、约13wt.%、约15wt.%、约20wt.%、多至约25wt.%、约30wt.%、约45wt.%、或在由前述值中的任意两个限定的任意范围内的硅含量,以及3wt.%、约5wt.%、约10wt.%、约20wt.%或更大、或在由前述值中的任意两个限定的任意范围内的羰基含量,例如约10wt.%-约45wt.%的硅含量以及3wt.%-约20wt.%的羰基含量,或者约15wt.%-约25wt.%的硅含量以及约5wt.%-约10wt.%的羰基含量。在一个示例性实施方案中,所述碳硅烷聚合物具有以道尔顿表示的大至5000、3500、2500、2000、1500、小至1000、800、500、或更小或在由前述值中的任意两个限定的任意范围内的重均分子量,例如1,000或更小,800-3500,800-2500,或800-1500。1.碳硅烷单体所述碳硅烷聚合物部分由碳硅烷单体组分形成。在一个示例性实施方案中,所述碳硅烷单体具有式:其中:x选自直链或支链的c1-c12烷基或c6-c14芳基,并且每个r是可水解的基团或不可水解的基团。在一个更具体的实施方案中,x选自直链c1-c12烷基。在一个甚至更具体的实施方案中,x选自甲基、乙基、苯基、二苯基、乙烯基(ethylene)和萘基。在又一更具体的实施方案中,x是乙基。示例性的可水解的基团包括c1-c12烷氧基、c1-c12烷硫基、c1-c12卤代烷氧基。示例性的不可水解的基团包括c1-c12烷基、苯基、芳基、乙烯基、丙烯酸酯基、环氧基和乙酰基。在一个更具体的实施方案中,每个r独立地选自c1-c12烷氧基,并且甚至更具体而言,每个r独立地选自甲氧基、乙氧基、异丙氧基、乙酰氧基、乙烯基、环氧基和乙酰基。在一个示例性实施方案中,每个r为乙氧基或甲氧基,并且在一个更具体的实施方案中,每个r为乙氧基。在一个示例性实施方案中,所述碳硅烷单体包括1,2-双(三乙氧基甲硅烷基)乙烷(“btse”)。btse具有式:。在一个示例性实施方案中,所述碳硅烷单体包括1,2-双(三乙氧基甲硅烷基)甲烷。1,2-双(三乙氧基甲硅烷基)甲烷具有式:。在一个示例性实施方案中,所述碳硅烷单体包括4,4-双(三乙氧基甲硅烷基)-1,1-联苯。4,4-双(三乙氧基甲硅烷基)-1,1-联苯具有式:。在一个示例性实施方案中,所述碳硅烷单体包括1,4-双(三乙氧基甲硅烷基)苯。1,4-双(三乙氧基甲硅烷基)苯具有式:。2.贡献羰基的单体所述碳硅烷聚合物部分由贡献羰基的单体形成。在一个示例性实施方案中,所述贡献羰基的单体包含选自丙烯酸类部分、羧基部分和酸酐部分的反应性部分。不希望受任何理论的束缚,但据信羰基更易于在氢气或氮气环境中还原,提高干蚀刻速率。还据信含羰基的部分对于胺型消解(digestion)溶液是更响应的,提高湿蚀刻速率。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的丙烯酸类单体:其中:y选自直链或支链的c1-c12烷基,r7、r8和r9各自是可水解的基团或不可水解的基团,并且r10、r11和r12各自是氢或取代的烃基。在一个更具体的实施方案中,y选自直链c1-c12烷基,并且甚至更具体而言,y是c1-c3烷基。在一个示例性实施方案中,y选自ch2、(ch2)2、(ch2)3、异丙基。在一个甚至更具体的实施方案中,y是c1或c2烷基,并且在又一更具体的实施方案中是c2烷基。示例性的可水解的基团包括c1-c12烷氧基、c1-c12烷硫基、c1-c12卤代烷氧基。示例性的不可水解的基团包括c1-c12烷基、苯基、芳基、乙烯基、丙烯酸酯基、环氧基和乙酰基。在一个更具体的实施方案中,r7、r8和r9各自独立地选自c1-c12烷氧基。在一个示例性实施方案中,r7、r8和r9各自独立地选自甲氧基和乙酰氧基。在一个示例性实施方案中,r7、r8和r9各自独立地选自甲氧基和乙氧基。在一个示例性实施方案中,r7、r8和r9各自是乙氧基。示例性的取代的烃基包括烷基、芳基、环氧基、缩醛基团、醚基团和芳基。在一个示例性实施方案中,r10、r11和r12各自选自氢或c1-c12烷基,并且甚至更具体而言,r10、r11和r12各自独立地选自氢或c1-c4烷基。在一个示例性实施方案中,r10、r11和r12各自为氢。在一个实施方案中,所述贡献羰基的单体是甲基丙烯酰氧基丙基三甲氧基硅烷。甲基丙烯酰氧基丙基三甲氧基硅烷为具有下式的丙烯酸类单体:。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的含羧基的单体:其中:y、r7、r8和r9如上所定义,并且r13是氢或取代的烃基。示例性的取代的烃基包括ch3。在另一示例性实施方案中,r13选自氢或c1-c12烷基、醚和环氧基,并且甚至更具体而言,r13选自氢或c1-c4烷基。在一个示例性实施方案中,r13选自甲基、乙基、丙基、异丙基、醚和环氧基。在一个示例性实施方案中,r13是氢。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的含酸酐的单体:其中:y、r7、r8和r9如上所定义,并且r14是氢或取代的烃基。示例性的取代的烃基包括ch3。在另一示例性实施方案中,r14选自氢或c1-c12烷基、醚和环氧基,甚至更具体而言,r14选自氢或c1-c4烷基。在一个示例性实施方案中,r14选自甲基、乙基、丙基、异丙基、醚和环氧基。在一个示例性实施方案中,r14是氢。c.其他组分除了所述碳硅烷聚合物之外,形成间隙填充或平坦化材料的所述组合物可以包含一种或多种任选的组分,例如交联促进剂、溶剂、发色体、催化剂、成孔剂和表面活性剂。还可以包含其他有机烷氧基硅烷单体。1.交联促进剂在一个实施方案中,所述组合物包含至少一种交联促进剂。示例性的交联促进剂包括氨基硅烷盐,例如apteos三氟甲磺酸盐、甘脲和通过诸如酸生成源热致酸生成剂和光致酸生成剂驱使的类似交联促进剂。在一个实施方案中,所述交联促进剂是具有下式的氨基硅烷盐:si(or)3(ch2)nnh3+(f3cso3)-其中n是1-10的整数,每个r独立地为c1-c20烷基。在一个更具体的实施方案中,所述交联促进剂是氨基丙基三乙基硅烷。示例性的氨基丙基盐是具有下式的apteos三氟甲磺酸盐:si(och2ch3)3(ch2)3nh3+(f3cso3)-。在一个实施方案中,按湿重计,所述交联促进剂占所述组合物的总重量的少至约0wt.%、约0.1wt.%、约0.25wt.%、约0.5wt.%、多至约1wt.%、约2wt.%、约5wt.%、约10wt.%、或在由前述值中的任意两个限定的任意范围内,例如0wt.%-约10wt.%,约0.1wt.%-约10wt.%,或约0.5wt.%-约1wt.%。2.溶剂在一个实施方案中,所述组合物包含至少一种溶剂。示例性的溶剂包括丙二醇单甲醚乙酸酯(pgmea)、醇诸如乙醇和异戊醇和水及其混合物。在一个实施方案中,所述溶剂包含平坦化增强剂。示例性的平坦化增强剂包括碳酸烷基酯,例如丙二醇碳酸酯(pc)。不希望受任何理论的束缚,但据信当被旋涂至基材时,所述丙二醇碳酸酯充当表面张力改性剂,其促进该溶液的平坦化效果。不希望受任何理论的束缚,但据信该溶剂混合物中的平坦化增强剂的效果与单体的选择无关。在一个实施方案中,所述至少一种溶剂包含第一溶剂和第二溶剂。示例性的第一溶剂包括pgmea和异戊醇。示例性的第二溶剂包括平坦化增强剂,例如丙二醇碳酸酯。在一个实施方案中,按湿重计,所述平坦化增强剂占所述组合物的总重量的少至约0wt.%、约2wt.%、约4wt.%、多至约5wt.%、约7wt.%、约7.1wt.%、约10wt.%、或在由前述值中的任意两个限定的任意范围内。在一个实施方案中,按湿重计,溶剂的总量占所述组合物的总重量的少至约0wt.%、约20wt.%、约40wt.%、多至约50wt.%、约60wt.%、约80wt.%、或在由前述值中的任意两个限定的任意范围内。3.发色体在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种发色体。示例性的发色体包括吸收248nm处的光的9-蒽羧基-烷基三烷氧基硅烷,例如9-蒽羧基-乙基三乙氧基硅烷(tesac)、9-蒽羧基-丙基三甲氧基硅烷和9-蒽羧基-丙基三乙氧基硅烷(actep)。其他示例性的发色体包括含苯基的硅烷,例如吸收193nm处的光的苯基三乙氧基硅烷(pteos)。其他示例性的发色体包括乙烯基teos和蒽发色体的萘类似物,例如出现在美国专利7,012,125中的那些,该专利的公开内容通过引用并入本文。示例性的发色体包括ah2006、ah2013、ah2015和ah2016,其结构式提供如下。ah2006:ah2013:ah2015:ah2016:。在一个实施方案中,基于包含单体的碳硅烷聚合物的总摩尔数,所述发色体占少至约3mol.%、约5mol.%、约10mol.%、多至约20mol%、约40mol.%、约60mol.%、或在由前述值中的任意两个限定的任意范围内,例如约3mol.%-约60mol.%,约5mol.%-约40mol.%,或约10mol.%-约20mol.%。在一个实施方案中,按干膜计,所述发色体占所述组合物的总重量的少至约3wt.%、约5wt.%、约10wt.%、约20wt.%、多至约25wt.%、约30wt.%、约35wt.%、约40wt.%、约60wt.%、或在由前述值中的任意两个限定的任意范围内,例如约3wt.%-约60wt.%,约5wt.%-约40wt.%,约10wt.%-约35wt.%,或约20wt.%-约30wt.%。4.催化剂在任一个上述实施方案的一个更具体的实施方案中,所述组合物还包含至少一种催化剂。示例性的催化剂包括四甲基硝酸铵(tman)和四甲基乙酸铵(tmaa)。另外的示例性催化剂可以见于美国专利8,053,159,该专利的公开内容通过引用整体并入本文。在一个实施方案中,按湿重计,所述催化剂占所述组合物的总重量的少至约0wt.%、约2wt.%、约4wt.%、多至约5wt.%、约7wt.%、约10wt.%、或在由前述值中的任意两个限定的任意范围内,例如约2wt.%-约10wt.%,约2wt.%-约7wt.%,约4wt.%-约7wt.%,或约5wt.%-约7wt.%。5.有机烷氧基硅烷单体在任一个上述实施方案的一个更具体的实施方案中,所述碳硅烷聚合物进一步由至少一种有机烷氧基硅烷单体形成。在一个甚至更具体的实施方案中,所述至少一种有机烷氧基硅烷单体选自甲基三甲氧基硅烷(mtmos)、甲基三乙氧基硅烷(mteos)、二甲基二乙氧基硅烷(dmdeos)、苯基三乙氧基硅烷(pteos)、二甲基二甲氧基硅烷、苯基三甲氧基硅烷、二苯基二乙氧基硅烷、二苯基二甲氧基硅烷和9-蒽羧基-烷基三烷氧基硅烷和前述物质的组合。在一个示例性实施方案中,所述有机烷氧基硅烷单体被并入所述碳硅烷聚合物中,并且更具体而言,被并入所述碳硅烷聚合物的主链中。在一个实施方案中,按湿重计,一种或多种有机烷氧基硅烷单体占所述组合物的总重量的小至约0wt.%、约20wt.%、约40wt.%、多至约50wt.%、约60wt.%、约80wt.%、或在由前述值中的任意两个限定的任意范围内,例如0wt.%-约80wt.%,约20wt.%-约60wt.%,或约40wt.%-约50wt.%。d.形成干膜的方法1.形成碳硅烷聚合物在一个实施方案中,所述碳硅烷聚合物通过使所述碳硅烷单体和所述贡献羰基的单体在溶剂溶液中反应以形成所述碳硅烷聚合物来形成。说明性的溶剂包括丙二醇甲醚乙酸酯(pgmea)、乙醇、水及其混合物。在一个实施方案中,所述碳硅烷聚合物通过催化的水解和缩合反应形成。在一个更具体的实施方案中,所述水解和缩合反应是酸催化反应。将酸(例如硝酸)加入所述碳硅烷单体、贡献羰基的单体以及任选的一种或多种其他组分例如发色体中以形成反应混合物。在一个实施方案中,将所述反应混合物加热以引发聚合反应。在一个实施方案中,将反应物加热至低至50℃、55℃、60℃、65℃、高至70℃、75℃、80℃、85℃、90℃的温度,持续少至1小时、1.5小时、2小时、多至2.5小时、3小时、3.5小时、4小时、或更长的时间。在一个实施方案中,在反应后,可以将所述混合物冷却,并且可以加入适当的淬灭剂(例如正丁醇)以停止反应。在冷却后,可以采用适当的溶剂,例如pgmea稀释所述混合物,并且可以加入一种或多种任选的组分,例如交联促进剂。在一些实施方案中,可以通过细孔过滤介质过滤所述混合物,以从材料中除去颗粒。2.形成干膜的方法在一个实施方案中,由包含碳硅烷聚合物的组合物形成膜。在一个实施方案中,通过旋涂将所述组合物施加于基材上。然后在低至约环境温度、约50℃、约100℃、约120℃、高至约180℃、约240ºc、约260℃、约300℃、或在由前述值中的任意两个限定的任意范围内、例如约50℃-约300℃、约100℃-约260℃、约120℃-约260℃、或约180℃-约240℃的温度下烘烤施加的组合物。将施加的组合物烘烤少至约10秒、约30秒、约1分钟、长至约5分钟、约10分钟、约15分钟、约60分钟、或在由前述值中的任意两个限定的任意范围内的时间,例如10秒-60分钟,1分钟-15分钟,或5分钟-10分钟。在一个示例性实施方案中,将施加的组合物在10℃下烘烤60秒,然后在被冷却至环境温度之前在240℃下、在氮气氛围中烘烤60秒。e.包含平坦化增强剂的组合物在一个实施方案中,提供包含硅源和至少一种溶剂的组合物,其中所述至少一种溶剂包含平坦化增强剂。示例性的硅源包括有机烷氧基硅烷、碳硅烷单体和贡献羰基的单体。在一个示例性实施方案中,所述硅源包括一种或多种具有以下通式的有机烷氧基硅烷:r1xsi(or2)y其中r1为烷基、烯基、芳基或芳烷基,并且x为0-2的整数,并且其中r2为烷基或酰基,并且y是1-4的整数。在一个实施方案中,所述硅源包括选自甲基三甲氧基硅烷(mtmos)、甲基三乙氧基硅烷(mteos)、二甲基二乙氧基硅烷(dmdeos)、苯基三乙氧基硅烷(pteos)、二甲基二甲氧基硅烷、苯基三甲氧基硅烷和前述物质的组合的有机烷氧基硅烷。在一个示例性实施方案中,所述硅源包括一种或多种具有以下通式的碳硅烷单体:其中:x选自直链或支化的c1-c12烷基或c6-c14芳基,并且每个r是可水解的基团或不可水解的基团。在一个更具体的实施方案中,x选自直链c1-c12烷基。在一个甚至更具体的实施方案中,x选自甲基、乙基、苯基、二苯基、乙烯基和萘基(naphyl)。在又一更具体的实施方案中,x是乙基。示例性的可水解的基团包括c1-c12烷氧基、c1-c12烷硫基、c1-c12卤代烷氧基。示例性的不可水解的基团包括c1-c12烷基、苯基、芳基、乙烯基、丙烯酸酯基、环氧基和乙酰基。在一个示例性实施方案中,所述硅源包括一种或多种选自1,2-双(三乙氧基甲硅烷基)乙烷(btse)、1,2-双(三乙氧基甲硅烷基)甲烷、4,4-双(三乙氧基甲硅烷基)-1,1-联苯和1,4-双(三乙氧基甲硅烷基)苯的碳硅烷单体。在一个示例性实施方案中,所述硅源包括一种或多种贡献羰基的单体。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的丙烯酸类单体:其中:y选自直链或支链的c1-c12烷基,r7、r8和r9各自是可水解的基团或不可水解的基团,并且r10、r11和r12各自是氢或取代的烃基。在一个示例性实施方案中,所述硅源包括甲基丙烯酰氧基丙基三甲氧基硅烷。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的含羧基的单体:其中:y、r7、r8和r9如上所定义,并且r13是氢或取代的烃基。在一个示例性实施方案中,所述贡献羰基的单体是具有下式的含酸酐的单体:其中:y、r7、r8和r9如上所定义,并且r14是氢或取代的烃基。示例性的溶剂包括丙二醇单甲醚乙酸酯(pgmea)、醇诸如乙醇和异戊醇和水及其混合物。在一个实施方案中,所述溶剂包含平坦化增强剂。示例性的平坦化增强剂包括碳酸烷基酯,例如丙二醇碳酸酯(pc)。不希望受任何理论的束缚,但据信当被旋涂至基材时,所述丙二醇碳酸酯充当表面张力改性剂,其促进该溶液的平坦化效果。不希望受任何理论的束缚,但据信该溶剂混合物中的平坦化增强剂的效果与单体的选择无关。在一个实施方案中,所述至少一种溶剂包含第一溶剂和平坦化增强剂。示例性的第一溶剂包括pgmea和异戊醇。示例性的平坦化增强剂包括丙二醇碳酸酯。在一个实施方案中,按湿重计,所述平坦化增强剂占所述组合物的总重量的少至约0wt.%、约2wt.%、约4wt.%、多至约5wt.%、约7wt.%、约7.1wt.%、约10wt.%、或在由前述值中的任意两个限定的任意范围内。在一个实施方案中,按湿重计,溶剂的总量占所述组合物的总重量的少至约0wt.%、约20wt.%、约40wt.%、多至约50wt.%、约60wt.%、约80wt.%、或在由前述值中的任意两个限定的任意范围内。实施例根据下面的实施例制备示例性聚合物。1.实施例#1:将300.1克的丙二醇单甲醚乙酸酯pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞(stopper)的设置于覆套(mantle)上的1l烧瓶中,并将得到的共混物搅拌10分钟。将355克的具有c14h34o6si2的分子式的单体1,2-双(三乙氧基甲硅烷基)乙烷加入至该共混物中,然后加入45克的0.008n硝酸。打开通向冷凝器的冷却水,并使该混合物在80℃下反应3小时。然后使反应混合物冷却。在67℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用约30wt.%-约80wt.%的pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将8500ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。2.实施例#2:将39.7克的9-蒽羧基-甲基三乙氧基硅烷(tesac)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,然后在连续搅拌下加入300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)直至tesac完全溶解。将141.84克的具有c14h34o6si2的分子式的单体1,2-双(三乙氧基甲硅烷基)乙烷连同36克的0.008n硝酸溶液一起加入至该共混物中。打开通向冷凝器的冷却水,并使该混合物在60℃下反应2小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将3400ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。3.实施例#3:将300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,并将得到的共混物搅拌10分钟。在连续搅拌下将141.84克的具有c14h34o6si2的分子式的单体1,2-双(三乙氧基甲硅烷基)乙烷和43克的苯基三乙氧基硅烷(pteos)加入至该共混物中,然后加入36克的0.008n硝酸。打开通向冷凝器的冷却水,并使该混合物在70℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将8500ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。4.实施例#4:将300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,并将得到的共混物搅拌10分钟。将340.56克的具有c13h32o6si2的分子式的单体双(三乙氧基甲硅烷基)甲烷加入至该共混物中,然后加入0.008n硝酸。酸溶液的量从45克变化至81克,产生具有720amu–1750amu的mw范围的均聚物。打开通向冷凝器的冷却水,并使该混合物在80℃下反应3小时。然后使反应混合物冷却。在67℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将3600ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。5.实施例#5:将300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,并将得到的共混物搅拌10分钟。将306.5克的具有c13h32o6si2的分子式的单体双(三乙氧基甲硅烷基)甲烷以及47.8克的具有c24h38o6si2的分子式的4,4-双(三乙氧基甲硅烷基)-1,1-联苯加入至该共混物中,然后加入0.008n硝酸。酸溶液的量从45克变化至81克,产生具有720amu–1750amu的mw范围的均聚物。打开通向冷凝器的冷却水,并使该混合物在60℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将3600ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。6.实施例#6:将300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,并将得到的共混物搅拌10分钟。将248.35克的3-甲基丙烯酰氧基丙基三甲氧基硅烷加入至该共混物中,然后加入36克的0.008n硝酸。打开通向冷凝器的冷却水,并使该混合物在80℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将8500ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性。7.实施例#7将300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,并将得到的共混物搅拌10分钟。将单体1,2-双(三乙氧基甲硅烷基)乙烷和具有分子式c10h22o4si的3-甲基丙烯酰氧基丙基三甲氧基硅烷加入至该共混物中。硅氧烷单体的量从283.67克的双(三乙氧基甲硅烷基)乙烷和49.67克的3-甲基丙烯酰氧基丙基三甲氧基硅烷变化至0克的3-甲基丙烯酰氧基丙基三甲氧基硅烷和248.35克的3-甲基丙烯酰氧基丙基三甲氧基硅烷。通过改变硅氧烷单体的量使硅的重量百分数从19.9wt.%变化至35.7wt.%。将36克的0.008n硝酸加入至该混合物中。打开通向冷凝器的冷却水,并使该混合物在60℃下反应2小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将8500ppm的apteos三氟甲磺酸盐加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。接着参考表1,使用实施例7的方法通过改变碳硅烷单体(btse)和含羰基的单体(3-甲基丙烯酰氧基丙基三甲氧基硅烷)的量来制备具有变化的硅含量的材料。对照材料不含含羰基的单体。在1500rpm下将每种材料浇铸于300mm晶片上并在130℃下烘烤60秒,随后在220℃下烘烤60秒。在以下溶剂中确定每个膜的蚀刻特性:pgmea,在室温下持续1分钟;2.38%tmah,在室温下持续1分钟;含水的碱性剥离剂clk-888,在室温下持续1分钟;clk-888,在30℃下持续1分钟;clk-888,在50℃下持续1分钟,和氢氧化铵,在40℃下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表1中。负值是由于膜膨胀而引起的。表1:实施例7的湿蚀刻数据材料pgmea,在rt下tmah,在rt下clk-888,在rt下clk-888,在30℃下clk-888,在50℃下nh4oh,在40℃下对照物(42.5wt.%si)-3%-1%-3%-2%100%0%35.7wt.%si-5%0%1%1%100%0%29.8wt.%si0%0%1%4%100%1%24.5wt.%si3%4%5%8%100%4%19.9wt.%si-2%-2%5%9%100%5%如表1中所示,在50℃下的clk-888中,每个膜在1分钟内被完全除去,并且所有的膜都耐pgmea(在室温下持续1分钟)。降低材料中的硅含量带来在室温和在30℃下的clk-888的剥离率的提高。8.实施例8:将39.7克的9-蒽羧基-甲基三乙氧基硅烷(tesac)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,然后在连续搅拌下加入300.1克的pgmea(ppt级)和600克的3a乙醇(不含甲苯)直至tesac完全溶解。将单体1,2-双(三乙氧基甲硅烷基)乙烷和具有分子式c10h22o4si的3-甲基丙烯酰氧基丙基三甲氧基硅烷加入至该共混物中。单体的量从88.65克的双(三乙氧基甲硅烷基)乙烷和37.25克的3-甲基丙烯酰氧基丙基三甲氧基硅烷变化至0克的1,2-双(三乙氧基甲硅烷基)乙烷和198.68克的3-甲基丙烯酰氧基丙基三甲氧基硅烷。通过改变硅氧烷单体的量使硅的重量百分数变化。将36克的0.008n硝酸加入至该混合物中。打开通向冷凝器的冷却水,并使该混合物在60℃下反应2小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将3400ppm的氨基丙基三乙氧基硅烷加入至最终的配制物中。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。接着参考表2,使用实施例8的方法通过改变碳硅烷单体(btse)和含羰基的单体(3-甲基丙烯酰氧基丙基三甲氧基硅烷)的量来制备具有变化的硅含量的材料。对照材料不含含羰基的单体。在1500rpm下将每种材料浇铸于300mm晶片上并在130℃下烘烤60秒,随后在240℃下烘烤60秒。在以下溶剂中确定每个膜的蚀刻特性:sc-1溶液(standardclean-1,按体积计包含1份的29%nh4oh水溶液、18份的30%h2o2水溶液和60份的di水),在70℃下持续1分钟;2.38%tmah,在室温下持续1分钟;含水的碱性剥离剂clk-888,在室温下持续1分钟;clk-888,在30℃下持续1分钟;和29%氢氧化铵,在40℃下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表2中。负值是由于膜膨胀而引起的。表2:实施例8的湿蚀刻数据材料sc-1,在60℃下tmah,在rt下clk-888,在rt下clk-888,在30℃下nh4oh,在40℃下对照物(31wt.%si)-2%0%-1%100%0%28.4wt.%si1%1%1%100%-3%23.8wt.%si1%1%1%100%7%19.6wt.%si4%4%100%100%5%15.8wt.%si6%11%100%100%7%如表2中所示,在30℃下的clk-888中,每个膜在1分钟内被完全除去。在温和的室温下的clk-888中的剥离率随着硅重量百分数的降低而增加。通过将硅含量从31wt.%降至23.8wt.%获得从0%至60%的去除率的增加,并且通过将硅含量进一步降至19.6wt.%或更低获得至100%去除率的增加。降低材料中的硅含量带来在室温和在30℃下的clk-888的剥离率的提高。在70℃下的sc-1中的平均蚀刻速率提供在下表3中。表3:实施例8的湿蚀刻速率材料烘烤条件平均蚀刻速率对照物(31wt.%si)140℃/220℃,每个温度下60秒-123.8wt.%si140℃/220℃,每个温度下60秒219.6wt.%si140℃/220℃,每个温度下60秒31如表3中所示,平均湿蚀刻速率随着硅含量的降低而增加。接着参考表4和图3和图4,其说明了对照物和20wt.%和24wt.%的硅材料的等离子体蚀刻数据,连同硅烷氧化物(silaneoxide)的等离子体蚀刻数据。图3图示说明了使用cf4/ar/o2的45/30/22组成在100mt、250w下在appliedmaterials(mxp)等离子体蚀刻工具中以a/分钟表示的蚀刻速率。图4图示说明了使用cf4/ar/chf3的30/500/30组成在300mt、800w下以a/分钟表示的蚀刻速率。表4:实施例8的等离子体蚀刻速率材料蚀刻速率(cf4/ar/o2)蚀刻速率(cf4/ar/chf3)对照物(31wt.%si)1262279923.8wt.%si4031133319.6wt.%si38331105如图3中图示说明的,cf4/ar/o2的等离子体蚀刻速率随着硅重量百分数的降低而增加。20wt.%硅材料是硅烷氧化物的蚀刻速率的5倍。然而,如图4中图示说明的,对于cf4/ar/chf3的等离子体蚀刻速率随着硅重量百分数的降低而降低。在图4中,较低的硅含量导致等离子体蚀刻速率的降低。接着参考表5,来自上表2的15.8wt.%的si样品的另外的样品,除了一组样品仅用pgmea稀释,而第二组样品用pgmea和丙二醇碳酸酯的共混物稀释。在两组样品上实施凝胶渗透色谱法。每个样品的数均分子量(mn)、重均分子量(mw)和多分散性(pd=mw/mn)提供在表5中。表5:实施例8的gpc结果材料mnmwpd15.8wt.%si,仅pgmea6457231.120515.8wt.%si,pgmea/pc共混物6557321.1165接着参考表6和表7,发现通过改变烘烤条件优化了表2的23.8wt.%硅材料和19.6wt.%硅材料的蚀刻特性。如上所述地制备另外的膜,但是根据表6或表7中给出的条件烘烤每种材料。在以下溶剂中确定每个膜的蚀刻特性:pgmea,在室温下持续1分钟;2.38%tmah,在室温下持续1分钟;clk-888,在室温下持续1分钟;sc-1溶液(standardclean-1,按体积计包含1份的29%nh4oh水溶液、18份的30%h2o2水溶液和60份的di水),在40℃下持续3分钟;和98%乙酸正丁酯,在室温下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表6和表7中。负值是由于膜膨胀而引起的。表6.仅用pgmea稀释的15.8wt.%si硅材料的其他湿蚀刻数据材料烘烤条件pgmea,在rt下tmah,在rt下clk-888,在rt下sc1,在40℃下nba15.8wt.%si,pgmea140℃/200℃,每个温度下60秒23%1%100%21%3%15.8wt.%si,pgmea140℃/210℃,每个温度下60秒15%8%100%11%1%15.8wt.%si,pgmea140℃/220℃,每个温度下60秒12%0%100%11%0%15.8wt.%si,pgmea140℃/230℃,每个温度下60秒8%1%100%6%0%15.8wt.%si,pgmea140℃/240℃,每个温度下60秒6%2%100%4%-2%15.8wt.%si,pgmea140℃/250℃,每个温度下60秒9%-2%100%2%0%如表6所示,每个膜在clk-888中被完全除去。观察到在pgmea中膜厚度减少,特别是对于在第二步骤中小于230℃的烘烤条件而言。表7:用pgmea/pc共混物稀释的15.8wt.%si硅材料的其他湿蚀刻数据材料烘烤条件pgmea,在rt下tmah,在rt下clk-888,在rt下sc1,在40℃下nba15.8wt.%si,pgmea/pc140℃/200℃,每个温度下60秒23%1%100%3%20%15.8wt.%si,pgmea/pc140℃/210℃,每个温度下60秒19%5%100%3%16%15.8wt.%si,pgmea/pc140℃/220℃,每个温度下60秒13%4%100%1%10%15.8wt.%si,pgmea/pc140℃/230℃,每个温度下60秒8%4%100%5%7%15.8wt.%si,pgmea/pc140℃/240℃,每个温度下60秒3%5%100%1%6%15.8wt.%si,pgmea/pc140℃/250℃,每个温度下60秒3%0%100%0%6%如表7所示,每个膜在clk-888中被完全除去。观察到在pgmea中膜厚度减少,特别是对于在第二步骤中小于约230℃或240℃的烘烤条件而言。接着参考表8,研究了表2的15.8wt.%硅材料的蚀刻特性。如上所述地制备另外的膜,但是将每种材料在140℃下烘烤60秒,随后在240℃下烘烤60秒。在以下溶剂中确定每个膜的蚀刻特性:sc-1溶液(standardclean-1,按体积计包含1份的29%nh4oh水溶液、18份的30%h2o2水溶液和60份的di水),在70℃下持续3分钟;pgmea,在室温下持续1分钟;2.38%tmah,在室温下持续1分钟;clk-888,在室温下持续1分钟;和98%乙酸正丁酯,在室温下持续1分钟;和29%氢氧化铵,在40℃下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表8中。表8:15.8wt.%硅材料的其他湿蚀刻数据材料sc1,在40℃下pgmea,在rt下tmah,在rt下clk-888,在rt下nbanh4oh15.8wt.%si4%4%1%100%1%1%如表8所示,每个膜在clk-888中被完全除去。经烘烤的膜耐pgmea、2.38%tmah和乙酸正丁酯。9.实施例9#在连续搅拌下将300.1克的丙二醇单甲醚乙酸酯pgmea(ppt级)和600克的3a乙醇(不含甲苯)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中。将不同量的1,2-双(三乙氧基甲硅烷基)乙烷、苯基三乙氧基硅烷和3-甲基丙烯酰氧基丙基三甲氧基硅烷加入至该共混物中,然后加入36克的0.008n硝酸。使反应混合物在70℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用pgmea(ppt级)将反应混合物稀释至目标膜厚度。在稀释后,将8500ppm的氨基丙基三乙氧基硅烷加入至最终的配制物中。将该溶液混合1小时以确保均匀性。接着参考表9,使用实施例9的方法通过改变碳硅烷单体(btse)和单体(tesac)的量来制备具有不同的硅含量的材料。对照材料不含tesac。在1500rpm下将每种材料浇铸于300mm晶片上并在130℃下烘烤60秒,随后在220℃下烘烤60秒。在以下溶剂中确定每个膜的蚀刻特性:pgmea,在室温下持续1分钟;2.38%tmah,在室温下持续1分钟;clk-888,在室温下持续1分钟;和clk-888,在30℃下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表9中。负值是由于膜膨胀而引起的。表9:实施例9的湿蚀刻数据材料pgmea在rt下tmah在rt下clk-888在rt下clk-888在30℃下对照物(36.2wt.%si)-1%-1%0%100%26.97wt.%si1%-4%37%100%20.4wt.%si-1%1%83%100%15.6wt.%si-2%4%100%100%如表9中所示,每个膜在30℃下的clk-888中,在1分钟内被完全除去,并且所有的膜都耐pgmea(在室温下持续1分钟)。所有的膜都耐2.3%tmah(在室温下),除了15.6wt.%si样品之外(其有4%的膜厚度被除去)。然而,通过将硅的重量百分数从36.2wt.%降至15.6wt.%使得在温和的室温下采用clk-888的剥离率从0%增至完全除去(100%)。接着参考表10,发现通过改变烘烤条件优化了15.6wt.%硅材料的蚀刻特性。如上所述地制备另外的膜,但是根据表10中给出的条件烘烤每种材料。在以下溶剂中确定每个膜的蚀刻特性:pgmea,在室温下持续1分钟;2.38%tmah,在室温下持续1分钟;和clk-888,在室温下持续1分钟。对于每种材料而言,在暴露之后的膜厚度的百分数变化呈现于表10中。负值是由于膜膨胀而引起的。表10:实施例9的其他湿蚀刻数据材料烘烤条件pgmea,在rt下tmah,在rt下clk-888,在rt下15.6wt.%si130℃/220℃,每个温度下60秒-2%4%100%15.6wt.%si130℃/220℃,每个温度下90秒2%3%100%15.6wt.%si130℃/230℃,每个温度下60秒0%1%100%15.6wt.%si130℃/230℃,每个温度下90秒-2%1%100%15.6wt.%si130℃/240℃,每个温度下60秒-8%1%86%15.6wt.%si130℃/240℃,每个温度下90秒1%1%50%如表10中所示,每个膜在30℃下的clk-888中,在1分钟内被完全除去。此外,通过增加烘烤温度改善了对室温下的2%tmah的抵抗性。此外,对于在130℃/220℃或130℃/230℃下烘烤的样品而言,在15.5wt.%达到100%的去除率。10.实施例10#将45.44克的9-蒽羧基-甲基三乙氧基硅烷(tesac)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,然后在连续搅拌下加入150.05克的异戊醇(iaa)和300克的2b乙醇直至tesac完全溶解。将124.8克的具有(c2h5o)4si的分子式的单体四乙氧基硅烷和77.7克的具有分子式ch3si(oc2h5)3的甲基三乙氧基硅烷,连同73.2克的0.008n硝酸溶液一起加入至该共混物中。打开通向冷凝器的冷却水,并使该混合物在60℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用异戊醇(iaa)稀释该反应混合物。根据上述方法制备类似的实施例,除了采用异戊醇(iaa)和丙二醇碳酸酯(pc)的溶剂共混物将反应混合物稀释至目标膜厚度。通过将100克的丙二醇碳酸酯加入至900克的异戊醇中来制备稀释溶剂共混物。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。将两种配制物涂覆于具有大平面(pad)样要素(14µm×45µm×60µm)的图案化晶片上,通过扫描电子显微镜(sem)分析确定整体平坦性。结果提供于表11中。表11:整体平坦性的比较材料整体平坦性iaa溶剂78.0iaa溶剂+pc平坦化增强剂47.6如表11所示,相比于采用不含平坦化增强剂的溶剂稀释的材料,采用包含平坦化增强剂的溶剂稀释的材料的平坦性改善了39%。11.实施例11#将39.7克的9-蒽羧基-甲基三乙氧基硅烷(tesac)加入至具有冷凝器、热电偶和瓶塞的设置于覆套上的1l烧瓶中,然后在连续搅拌下加入150.05克的丙二醇单甲醚乙酸酯pgmea(ppt级)和300克的3a乙醇(不含甲苯)直至tesac完全溶解。将17.7克的1,2-双(三乙氧基甲硅烷基)乙烷和86.9克的具有分子式c10h22o4si的3-甲基丙烯酰氧基丙基三甲氧基硅烷,连同36克的0.008n硝酸溶液一起加入至该共混物中。打开通向冷凝器的冷却水,并使该混合物在60℃下反应3小时。然后使反应混合物冷却。在57℃下,通过加入44.2克的正丁醇淬灭反应。使反应混合物冷却至室温,并在该温度下保持过夜。然后用丙二醇单甲醚乙酸酯pgmea(ppt级)稀释反应混合物。根据上述方法制备类似的实施例,除了采用丙二醇单甲醚乙酸酯pgmea(ppt级)和丙二醇碳酸酯(pc)的溶剂共混物将反应混合物稀释至目标膜厚度。通过将100克的丙二醇碳酸酯加入至900克的pgmea(ppt级)中来制备稀释溶剂共混物。将该溶液混合1小时以确保均匀性,然后通过细孔过滤介质过滤该溶液,以从材料中除去颗粒。将两种配制物涂覆于具有大平面样要素(14µm×45µm×60µm)的图案化晶片上,通过扫描电子显微镜(sem)分析确定整体平坦性。结果提供于表12中。表12:整体平坦性的比较材料整体平坦性pgmea溶剂14.5pgmea溶剂+pc平坦化增强剂7.5如表11所示,相比于采用不含平坦化增强剂的溶剂稀释的材料,采用包含平坦化增强剂的溶剂稀释的材料的平坦性改善了50%。尽管本发明已经被描述为具有示例性的设计,但是在本公开内容的精神和范围内可以进一步修改本发明。因此,本申请旨在使用本发明的基本原理涵盖本发明的任何变化、用途或修改。此外,本申请旨在涵盖出于本发明所涉及的领域中的已知或惯常的实践并且落入所附权利要求的范围的与本公开内容的这样的偏离。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种阻燃耐火硅聚合物组合物的制作方法与工艺
- 用水性分散体中的聚合物浸渍天然纤维的方法和所述纤维在复合材料中的用途与流程
- 细水性聚合物分散体用于浸渍天然纤维的用途的制作方法与工艺
- 水溶性聚合物水处理剂生产用管式合成系统的制作方法与工艺
- 非硫化硅烷改性聚合物锌粉膏及其用于悬索桥主缆的复合防护结构的制作方法与工艺
- 一种水性聚合物异氰酸酯木材胶粘剂及其制备方法与流程
- 一种盐增黏水溶性两亲聚合物驱油剂的制作方法与工艺
- 核壳型聚合物粒子、水性聚合物乳液、毛发化妆品组合物的制造方法与工艺
- 核壳型聚合物粒子、水性聚合物乳液、毛发化妆品组合物的制造方法与工艺
- 一系列基于莱啉基化合物的水溶性聚合物的制备及其作为光热试剂在生物医学的应用的制造方法与工艺
- 星形聚合物接枝多肽和聚乙烯亚胺的基因载体及制备方法与流程
- 一种木质纤维接枝丙烯酰胺聚合物及其制备方法与流程
- 含橡胶接枝聚合物粉体、以及含有其的太阳能电池用密封材料和夹层玻璃用中间膜的制作方法与工艺
- 附接到可降解的聚合物区的支撑件的制作方法与工艺
- 接枝聚合物、树脂着色物、该树脂着色物的制造方法和包含该树脂着色物的树脂组合物与流程
- 接枝聚合物、树脂着色物、该树脂着色物的制造方法和包含该树脂着色物的树脂组合物与流程
- 角蛋白接枝水性聚氨酯聚合物皮革复鞣填充材料的制备方法与流程
- 一种高接枝率聚烯烃接枝物的制备方法及该高接枝率聚烯烃接枝物的应用与流程
- 一种高抗冲聚丙烯接枝聚合物及其制备方法与流程
- 非硫化硅烷改性聚合物锌粉膏及其用于悬索桥主缆的复合防护结构的制作方法与工艺