一种碳化钼纳米片及其制备方法和应用与流程
本发明涉及碳化钼制备方法,尤其是一种碳化钼纳米片及其制备方法和应用。
背景技术:
氢能的利用是传统化石能源是佳的可替代绿色能源,而氢能的利用取决于制氢技术的发展。电解水制氢技术为行业誉为“最理想的工业制氢方法”,该技术的最核心问题是高效、稳定、廉洁的制备电催化剂的开发。目前,铂基催化剂是最为有效的电催化剂,但其高昂的成本和低存储量严重制约了该类催化剂的广泛应用。
碳化钼(mo2c)由于具有类贵金属性质、耐焙烧、耐硫耐氮、高熔点等优点,在催化领域中得到了广泛的关注,成为最具有潜力代替贵金属pt、pd的催化材料之一。目前,碳化钼的制备方法主要有化学气相沉积法、程序升温还原法和碳热氢还原法。化学气相沉积法可制备出高比表面、高质量的薄膜mo2c催化剂,但该方法的沉积效率比较低。程序升温还原法可制备纳米mo2c催化剂,同时具有较高的活性,但该方法需要复杂和严格的合成条件,可控性较差。碳热氢还原法通过控制活性组分和温度可合成高比表面积、晶粒大小可控的高性能mo2c催化剂。原料方面,碳源是合成高性能mo2c的关键之一,不同的碳源可合成不同晶体结构的mo2c催化剂。目前,主要采用的碳源有乙烷、甲烷、正庚烷和正丁烷等气体,碳源通常需要与氢气混合以实现碳化获得mo2c催化剂,合成工艺较为复杂。同时,现阶段mo2c的合成通过在较高的温度下进行,如以20%ch4~80%h2混合气为碳源,其反应需要在700~900℃,当时反应温度较低时,会导致碳化不完全。较高的合成温度会不可避免造成mo2c的烧结与团聚,引起催化剂的比表面积减小,严重影响了催化剂活性的发挥。
技术实现要素:
本发明的目的是为了克服现有的碳化钼合成温度高、产物团聚的问题,提供一种碳化钼纳米片及其制备方法和应用。
具体方案如下:
一种碳化钼纳米片的制备方法,包括以下步骤:
步骤1:将钼源、硝酸铵、甘氨酸混合均匀,之后加热到160~180℃,得到混合物1;
步骤2:向步骤1得到的混合物1中加入葡萄糖,之后加热到230~280℃,获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入氢气氛围下加热到450~550℃,降温得到碳化钼纳米片。
进一步的,步骤1中所述钼源为仲钼酸铵、四钼酸铵或二钼酸铵中至少一种;
任选的,步骤1中所述钼源中钼的摩尔的量:所述硝酸铵的摩尔的量+甘氨酸的摩尔的量=0.5~1.0:2.2~3.2;
任选的,步骤1中所述硝酸铵的质量:所述甘氨酸的质量=0.8~3.2。
进一步的,步骤1包括:1a将钼源、硝酸铵、甘氨酸混合后加入去离子水,搅拌至溶解;1b加热溶液到165~175℃,保温3~6分钟,得到混合物1。
进一步的,步骤2中所述葡萄糖与所述钼源中的钼的质量比为0.8~1.0:1.6~2.8;
任选的,步骤2中葡萄糖配制成浓度为0.1~0.3g/ml葡萄糖水溶液,向步骤1得到的混合物1中加入10~50ml的所述葡萄糖水溶液。
进一步的,步骤2中所述加热的温度为240~270℃,保温时间为10~20分钟。
进一步的,步骤3中所述氢气氛围中h2的流量为100~500ml/min。
进一步的,步骤3中所述加热的温度为460~500℃,保温时间为1~3小时。
本发明还保护所述的碳化钼纳米片的制备方法制备得到的碳化钼纳米片。
进一步的,所述碳化钼纳米片具有晶态结构,呈不规则片状,厚度为10~30nm。
本发明还保护所述的碳化钼纳米片在电催化制氢中的应用。
有益效果:
本发明中,所述的碳化钼纳米片采用葡萄糖作为碳源,在反应过程中,葡萄糖分散并受热形成无定型态的纳米碳片,作为碳化钼形成的结构模板,是决定碳化钼呈现片状纳米结构的关键之一。
再则,本发明利用硝酸铵和甘氨酸为反应助剂,不仅有效降低了碳化钼生成过程中的温度,实现了中温(450~550℃)完成碳氢还原合成碳化钼纳米片,而且还借助硝酸铵和甘氨酸反应过程中释放的大量还原性气体,形成包含非晶态氧化钼和无定型纳米碳片的前驱体。
总之,本发明所述的碳化钼纳米片制备方法加热温度低,便于操作和推广,所制得的碳化钼不会团聚,呈现不规则片状,比表面积大,在电催化制氢中的应用中具有优秀的催化性能。
附图说明
为了更清楚地说明本发明的技术方案,下面将对附图作简单的介绍,显而易见地,下面描述中的附图仅仅涉及本发明的一些实施例,而非对本发明的限制。
图1是本发明实施例1提供的前驱体的xrd衍射谱;
图2是本发明实施例1提供的前驱体的扫描电子显微镜照片;
图3是本发明实施例1提供的碳化钼纳米片的xrd衍射谱;
图4是本发明实施例1提供的碳化钼纳米片的扫描电子显微镜照片;
图5是本发明对比例1提供的致密固体的xrd衍射谱;
图6是本发明对比例1提供的对比样1的xrd衍射谱;
图7是本发明对比例1提供的对比样1的扫描电子显微镜照片;
图8是本发明对比例2提供的蓬松固体的xrd衍射谱;
图9是本发明对比例2提供的蓬松固体的扫描电子显微镜照片;
图10是本发明对比例2提供的对比样2的xrd衍射谱;
图11是本发明对比例2提供的对比样2的扫描电子显微镜照片。
具体实施方式
本发明中使用的术语的定义具有本领域所公知的定义和含义。
本发明提供的碳化钼纳米片制备方法中,钼源优选为仲钼酸铵,其与去离子水混合形成溶液,分散均一,有利于控制后续反应的速度,形成厚度均一的产物。
本发明提供的碳化钼纳米片制备方法中,所述钼源中的钼的摩尔的量:(所述硝酸铵的摩尔的量+所述甘氨酸摩尔的量)=0.5~1.0:2.2~3.2,优选0.7:2.5~2.9,例如0.7:2.7,小于该比例(0.5~1.0:2.2~3.2),硝酸铵+甘氨酸太多,反应过剧烈,产生的温度过高,容易使生成的前驱体成烧结粘连状态,无法完全形成非晶态结构和纳米片状结构,同时,相对钼源太少,产物的产量也低了。大于该比例,硝酸铵+甘氨酸太少,无法将钼源分解为氧化物。其中所述硝酸铵的质量:所述甘氨酸的质量=0.8~3.2,优选1.3~2.5:,更优选1.8~2.3,例如2.2,两者反应遵守化学计量比原则,小于或大于该比例均无法达到分解钼源所需的温度,同时,反应释放的气体量也不够形成纳米片结构。硝酸铵与甘氨酸发生氧化还原反应,反应放热,产生大量还原性气体,不仅作用于葡萄糖的还原过程,而且促使钼源形成包含非晶态氧化钼的前驱体。葡萄糖与所述钼源中钼的质量比为0.8~1.0:1.6~2.8,如果葡萄糖过量,则会形成过量的无定型纳米碳片,在碳氢还原过程中无法完全反应消除,会导致产物mo2c中残留较多的碳。
本发明提供的碳化钼纳米片制备方法中,步骤1加热到160~180℃,得到混合物1,优选165~175℃,保温3~6分钟,更优选为170℃,保温5分钟,通过加热使得钼源形成包含非晶态氧化钼的前驱体。
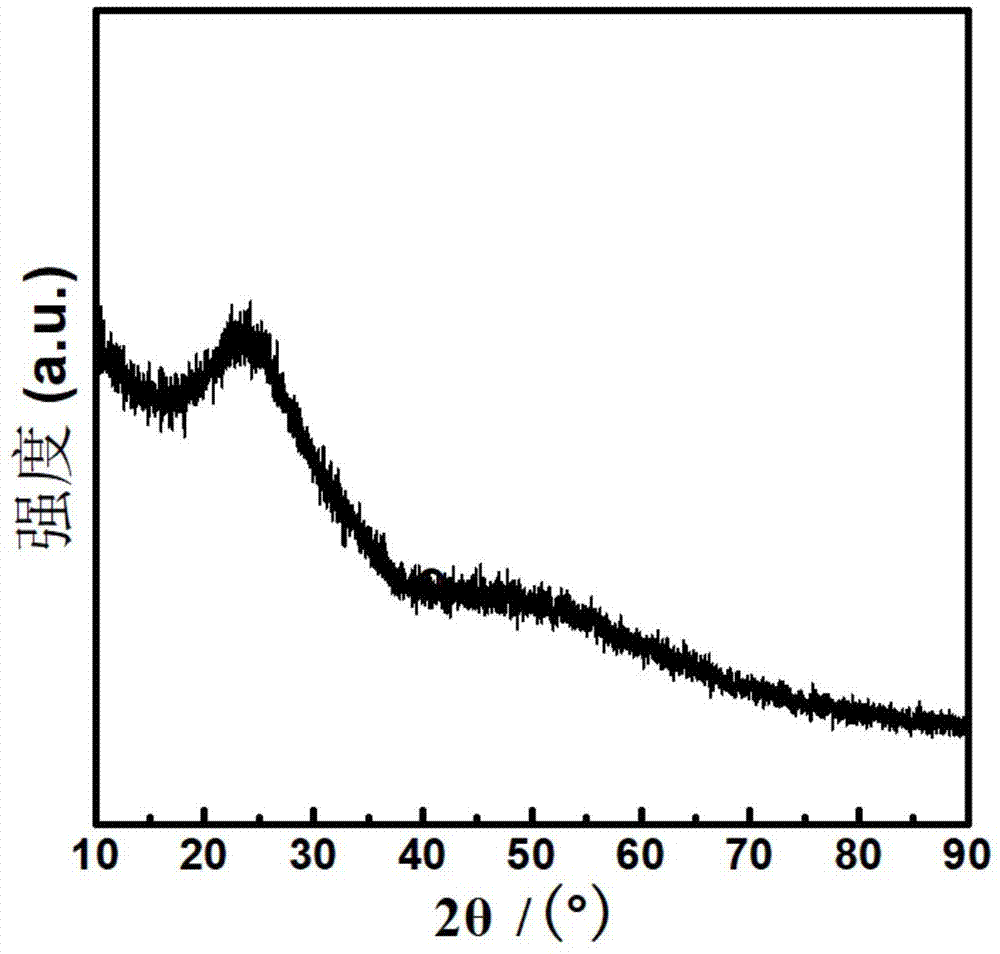
本发明提供的碳化钼纳米片制备方法中,步骤2加热到230~280℃,获得蓬松固体,优选为240~270℃,保温时间为10~20分钟,更优选为250℃,保温15分钟,该步获得蓬松固体为非晶态的氧化钼和无定型纳米碳片的混合物,通过其xrd衍射谱,可知,其在衍射角为23°形成明显的无定型态的衍射峰,表明其为由非晶态物质组成。
本发明提供的碳化钼纳米片制备方法中,步骤3加热到450~550℃,优选为480~530℃,保温时间为1~3小时,更优选为500℃,保温2小时,该反应需要在流量为100~500ml/min的氢气氛围下进行,以保证碳氢反应的进行,流量过小,碳氢反应不足,无法完全形成mo2c,流量达大则造成浪费。需要说明的是,所述氢气氛围可以是纯氢气,也可以是氢气与其他的气体混合,例如氢气与氮气的混合、或者氢气与惰性气体的混合,原则上只要含有氢气的气体都可以,对温度没有影响,但对氢气流量有影响,例如氢/氮混合气体,其气体流量必须足够大以保证氢气足够量,优选使用纯氢气,流量为100~500ml/min。
本发明制备的碳化钼纳米片可用于替代贵金属pt、pd作为催化剂,尤其是在电催化制氢中。通过对产品的成分和形貌分析可以看出,所制得的碳化钼纳米片不会团聚,呈现不规则片状,比表面积大,具有较好的结构优势,能够发挥出较好的催化性能。
本发明对各原料的来源没有特别的限定,可以通过商购得到,也可以按照现有的各种方法制备得到,原料优选为分析纯等级。此外,还需要说明的是,本发明对加热的实现的方式没有特别的限定,只要能够使反应体系处于稳定的温度范围即可。例如,马弗炉,管式炉。此外,保持体系在氢气氛围下,可以是通入氢气,也可以是氢气与其他惰性气体的混合,例如氢气与氮气混合,对此本领域技术人员均能知悉,在此不作赘述。
下面将更详细地描述本发明的优选实施方式。虽然以下描述了本发明的优选实施方式,然而应该理解,可以以各种形式实现本发明而不应被这里阐述的实施方式所限制。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。在下面的实施例中,如未明确说明,“%”均指重量百分比,度均指℃。
以下使用的测试方法包括:
xrd测试:通过x射线衍射仪(rigakud/max-rb12,xrd)测定产物的物相,测试条件为cu靶,kα(λ=0.1541nm)
扫描电子显微镜:采用场发射扫描电子显微镜(feiquantafeg450)对产物进行显微形貌观察。
实施例1
按照以下步骤制备碳化钼纳米片:
步骤1:将18.5克仲钼酸铵、20克硝酸铵、12克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在160℃加热5分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入30ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入240度的加热炉中加热12分钟,加热结束后获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入管式炉中,在氢气流量为200ml/min的流动氢气下,500℃碳氢还原1.5小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到碳化钼纳米片。
对步骤2中制得的蓬松固体、步骤3制得的碳化钼纳米片分别进行xrd测试和电镜测试,结果见图1~图4。
从图1可知,蓬松固体在衍射角为23°形成明显的无定型态的衍射峰,表明蓬松固体由非晶态物质组成,其组成为非晶态的氧化钼和无定型纳米碳片的混合物。图2为蓬松固体的扫描电子显微镜照片,从图2可知,蓬松固体成纳米片状结构,未发现明显的晶态相,这与xrd的结果相符。
图3是碳化钼纳米片的xrd衍射谱,从图3可知,碳化钼纳米片为晶态结构,图4是碳化钼的扫描电子显微镜照片,可以看到其为不规则的片状,片层厚度约15nm,具有较大的比表面积。
实施例2
步骤1:将11.2克四钼酸铵、18克硝酸铵、9克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在170℃加热4分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入20ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入250度的加热炉中加热10分钟,加热结束后获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入管式炉中,在氢气流量为150ml/min的流动氢气下,450℃碳氢还原2小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到碳化钼纳米片。经扫描电子显微镜测试,其为不规则的片状,片层厚度尺寸约20nm。
实施例3
步骤1:将23.6克二钼酸铵、22克硝酸铵、13克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在165℃加热5分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入20ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入230度的加热炉中加热15分钟,加热结束后获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入管式炉中,在氢气流量为150ml/min的流动氢气下,550℃碳氢还原1小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到碳化钼纳米片。经扫描电子显微镜测试,其为不规则的片状,片层厚度尺寸约25nm。
实施例4
步骤1:将19.3克仲钼酸铵、20克硝酸铵、25克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在180℃加热3分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入23ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入240度的加热炉中加热10分钟,加热结束后获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入管式炉中,在氢气流量为500ml/min的流动氢气下,460℃碳氢还原3小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到碳化钼纳米片。
实施例5
步骤1:将26.3克仲钼酸铵、20克硝酸铵、6.3克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在170℃加热6分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入26ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入270度的加热炉中加热10分钟,加热结束后获得蓬松固体;
步骤3:将步骤2得到的蓬松固体放入管式炉中,在氢气流量为100ml/min的流动氢气下,500℃碳氢还原1小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到碳化钼纳米片。
对比例1
步骤1:将18.5克仲钼酸铵放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成溶液,将盛有上述溶液的烧杯放入马弗炉中,在160℃加热5分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将葡萄糖加入离子水配制成浓度为0.2g/ml葡萄糖溶液,并步骤1加热结束后的溶液中加入30ml的葡萄糖溶液,并进行搅拌形成混合溶液,将混合溶液放入240度的加热炉中加热12分钟,加热结束后获得的是致密固体而非蓬松态粉末;
步骤3:将步骤2得到的致密固体放入管式炉中,在氢气流量为200ml/min的流动氢气下,500℃碳氢还原1.5小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到对比样1。
对步骤2中制得的致密固体、步骤3制得的对比样分别进行xrd测试和电镜测试,结果见图5~图7。
从图5可知,致密固体仍形成明显的无定型态的衍射峰,但同时出现了氧化钼(二氧化钼、三氧化钼)的衍射峰,表明氧化钼已变成结晶态相,无法形成非晶态的氧化钼,这将导致在后续的碳氢还原步骤中无法顺利合成碳化钼。
图6是步骤3制得的对比样1的xrd衍射谱,从图6可知,对比样1由主相二氧化钼和少量碳化钼组成,表明无法成功合成碳化钼相。图7是比对样1的扫描电子显微镜照片,可以看到其形貌为大尺寸颗粒状,无法形成纳米片状形貌结构。
对比例2
步骤1:将18.5克仲钼酸铵、20克硝酸铵、12克甘氨酸同时放入1000ml的烧杯中,并加入100ml去离子水,搅拌到完全溶解形成混合溶液,将盛有上述混合溶液的烧杯放入马弗炉中,在160℃加热5分钟,加热过程中保持与空气充分接触,同时充分进行搅拌;
步骤2:将柠檬酸配制成浓度为0.2g/ml柠檬酸溶液,并步骤1加热结束后的溶液中加入30ml的柠檬酸溶液,并进行搅拌形成混合溶液,将混合溶液放入240度的加热炉中加热12分钟,加热结束后获得较为蓬松固体粉末;
步骤3:将步骤2得到的较蓬松固体粉末放入管式炉中,在氢气流量为200ml/min的流动氢气下,500℃碳氢还原1.5小时,碳氢还原结束后仍保温流动氢气状态,待炉温降至室温后将粉末取出,得到对比样2。
对步骤2中制得的致密固体、步骤3制得的对比样分别进行xrd测试和电镜测试,结果见图8~图11。
从图8可知,蓬松固体粉末形成明显的无定型态的衍射峰,表明蓬松固体由非晶态物质组成,其组成为非晶态的氧化钼和无定型纳米碳片的混合物。图9为较蓬松固体粉末的扫描电子显微镜照片,从图9可知,较蓬松固体粉末为多孔片状结构,厚度约2~4微米,无法形成纳米片状结构,这与图2有明显区别。
图10是比对样2的xrd衍射谱,从图10可知,对比样2由主相碳化钼和次相二氧化钼组成,表明无法完全合成碳化钼相。图11是比对样2的扫描电子显微镜照片,从图11可知,对比样2仍保持碳氢还原前的形貌,即多孔片状结构,无法形成纳米片状结构,这与图4有明显区别。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种纳米粉体工业生产系统的制作方法
- 一种高红外辐射粉体及其制备方法
- 一种铈锌共掺NiFe<sub>2</sub>O<sub>4</sub>纳米粉体及其制备方法
- 一种纳米Al<sub>2</sub>O<sub>3</sub>/Er<sub>3</sub>Al<sub>5</sub>O<sub>12</sub>/ZrO<sub>2</sub>复合粉体材料的制备方法
- 一种β?氢氧化镍纳米片的制备方法
- 一种硫化亚铜纳米玫瑰花的制备方法
- 一种球形氧化钇粉体的制备方法
- 一种亚微米级碳化硼粉体及其制备方法和用图
- 一种B<sub>4</sub>C纳米片的制备方法及B<sub>4</sub>C纳米片的制作方法
- 氮化铝粉体的制备方法