一种植物等位基因不平衡表达检测方法与流程
本发明涉及基因表达检测领域,尤其涉及一种植物等位基因不平衡表达检测方法。
背景技术:
等位基因(allele又作allelomorph)一般指位于一对同源染色体的相同位置上控制着相对性状的一对基因。
等位基因的不平衡表达(allelic expression imbalance,AEI)为同一个细胞内,每个基因通常有2个拷贝,由于顺式作用使基因的2个拷贝的表达比例偏离了1:l。等位基因的不平衡表达现象普遍存在,除了遗传印迹基因的绝对不平衡表达外,有相当数量的基因在部分个体、同一个体不同时空上存在AEI。并与基因组一些特定区域的多态位点相关。
目前,常用的等位基因不平衡表达检测主要有基于电泳技术采用限制性片段长度多态、单链构象多态性技术、测序法、表达谱芯片法以及SNaPshot技术,测序法和SNaPshot价格比较昂贵,且须以相关的大型仪器平台作为支撑,实验操作灵活性差。
技术实现要素:
有鉴于此,本发明的目的在于提供一种等位基因不平衡表达检测方法,以解决现有技术中等位基因不平衡表达检测成本高、准确性差的问题。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种植物等位基因不平衡表达检测的方法,包括以下步骤:
1)比较候选基因序列,筛选出标记等位基因的SNPs位点;
2)根据所述步骤1)筛选到的SNPs位点设计qPCR特异引物和简并引物,所述qPCR特异引物包括正向引物和反向引物,对正向引物或反向引物3'末端与SNP位点3'末端互补的核苷酸进行LNA修饰;
3)用所述qPCR特异引物和简并引物分别对待测样本的cDNA进行PCR扩增,获得扩增产物;在PCR扩增过程中检测扩增曲线,在PCR扩增结束后检测扩增产物的熔解曲线;
4)若扩增产物的熔解曲线为单峰,根据PCR扩增的Ct值计算得到待测样本等位基因表达特异性;
所述qPCR特异引物包括正向引物和反向引物,对引物3'末端与SNP位点3'末端互补的核苷酸进行LNA修饰。
优选的,步骤3)中所述待测样本为林木树种。
优选的,所述待测样本为毛果杨或巨桉。
优选的,当待测样品为毛果杨时,所述SNP位点位于热激转录因子基因2上。
优选的,所述qPCR特异引物包括正向引物SEQ ID NO.1、SEQ ID NO.2和反向引物SEQ ID NO.4;所述SEQ ID NO.1和SEQ ID NO.2的3'末端上的倒数第二个核苷酸进行LNA修饰;所述简并引物包括正向简并引物SEQ ID NO.3和反向引物SEQ ID NO.4。
优选的,当待测样品为巨桉时,所述SNP位点位于MYB转录因子基因上。
优选的,所述qPCR特异引物包括正向引物SEQ ID NO.5、SEQ ID NO.6和反向引物SEQ ID NO.8;所述SEQ ID NO.5和SEQ ID NO.6的3'末端上的倒数第二个核苷酸进行LNA修饰;所述简并引物包括正向简并引物SEQ ID NO.7和反向引物SEQ ID NO.8。
优选的,所述待测样本cDNA的PCR扩增程序为:预变性94℃3min;变性94℃10s,退火62℃30s,延伸72℃40s;40个循环。
优选的,所述待测样本cDNA的PCR扩增体系包括:20ng/μL的待测样品cDNA,1μL;SYBR Premix Ex TaqTM(2×),10μL;10μM的正向引物0.5μL;10μM的反向引物0.5μL;双蒸水8μL。
优选的,根据权利要求1所述的方法,所述待测样本等位基因表达特异性计算的公式为2-ΔCt,ΔCt=候选等位基因基因型R的Ct值-候选等位基因基因型R’的Ct值;所述基因型R为A、T、C和G中的一种,所述基因型R’为A、T、C和G中除R以外的三种中的一种。
本发明的有益效果:本发明所提供的方法,采用LNA锁核酸修饰的引物对SNP位点进行qPCR扩增,得到扩增产物的熔解曲线,当熔解曲线为单峰时,以PCR扩增的Ct值计算等位基因不平衡表达特异性水平。本发明提供的方法采用LNA锁核酸修饰引物,可提高退火温度,增强碱基配对的特异性,能够极大地降低错配发生的概率,增加了检测结果的精确性和可靠性,并且所述方法仅仅采用实时荧光定量PCR仪器即可完成整个技术方案,简化了实验配套条件,显著地降低了实验成本。
附图说明
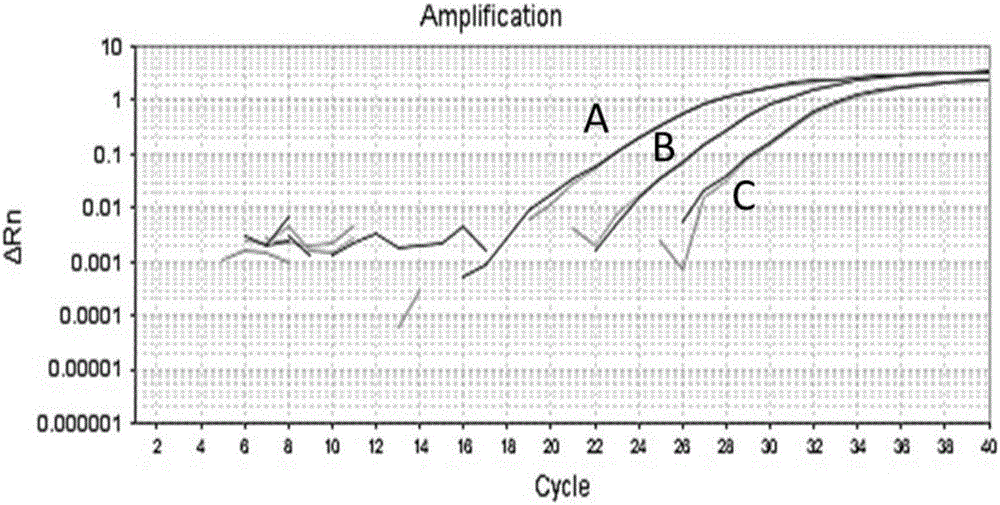
图1为毛果杨热击转录因子基因2的等位基因扩增曲线;
图2为巨桉MYB转录因子基因的等位基因扩增曲线。
具体实施方式
本发明提供了一种植物等位基因不平衡表达检测方法,包括以下步骤:1)比较候选基因序列,筛选标记等位基因的SNPs位点;2)根据筛选到的SNPs位点设计qPCR特异引物和简并引物;3)用qPCR特异引物和简并引物分别对待测样本的cDNA进行PCR扩增,获得扩增产物;4)若扩增产物的熔解曲线为单峰,根据PCR扩增的Ct值计算待测样本等位基因表达特异性;所述qPCR特异引物包括正向引物和反向引物,对引物3'末端与SNP位点3'末端互补的核苷酸进行LNA修饰。
本发明提供的检测方法适用于植物等位基因不平衡表达检测,优选的待测样本为林木树种,更优选的待测样本为毛果杨或巨桉。
在本发明中,比较候选基因序列,筛选出标记等位基因SNPs位点时针对物种的种类选择以下两种方法:针对模式物种,通过物种重测序数据库进行候选基因SNPs位点筛选,所述重测序数据库优选的为Phytozome数据库,网址为https://phytozome.jgi.doe.gov/;具体的在利用重测序数据库筛选SNPs位点时,筛选的SNPs位点满足以下条件:筛选的SNPs位点在群体中的频率大于5%,并且该位点在个体中为杂合状态。
对于非模式物种,通过候选基因cDNA克隆进行等位基因SNPs位点筛选,具体的在候选基因cDNA克隆进行等位基因SNPs位点筛选时,至少进行8个克隆测序,等位基因SNPs位点筛选成功率达到99.8%以上。所述候选基因cDNA克隆和筛选的方法采用本领域常规的基因克隆和筛选的方法即可,在本发明具体的包括以下步骤:根据候选基因保守序列设计引物,以cDNA为模板,扩增相应的基因片段。使用Takara公司的PMD-18T载体进行重组质粒的连接,连接体系为:5μL Solution I,,1μLpMD-18T Vector(50ng/μL),2μL PCR产物,12μL双蒸水,16℃连接过夜。然后进行重组质粒的转化:1)取5uL上述连接产物加入到50uL大肠杆菌TOP10感受细胞中,混匀,冰浴30min;2)42℃水浴热激90s后冰浴3min;3)加入200uL LB液体培养基,混匀。在37℃培养箱中150rpm培养30-60min;4)取100uL培养液,涂布于含Amp50mg/L的LB固体培养基上。37℃倒置培养过夜。最后,从平板上分别挑取10个单菌落于20μL液体LB培养基汇中。取单菌落进行PCR鉴定。对阳性菌落测序完成后,根据获得的基因序列设计5'与3'RACE引物,进行RACE PCR扩增,从而获得全长基因序列。
在本发明中,所述筛选到的标记等位基因SNPs位点优选的位于候选基因3'端的不保守区域,用于和基因家族中不同的成员进行区分。在本发明中,当待测样本为毛果杨时,筛选到的SNP位点优选的位于热激转录因子基因2(编号为Potri.001G108100)上;当待测样品为巨桉时,筛选到的SNP位点优选的位于MYB转录因子基因(编号为Eucgr.K02470)上。
本发明在筛选获得标记等位基因SNPs位点后,根据筛选到的SNPs位点设计qPCR特异引物和简并引物。所述qPCR特异引物包括正向引物和反向引物,对正向引物或反向引物3'末端与SNP位点3'末端互补的核苷酸进行LNA修饰。具体的在本发明中,所述正向引物或反向引物的3'末端倒数第一个或倒数第二个碱基与标记等位基因的SNPs位点配对,并在进行引物合成时将此碱基所在的核苷酸的糖环进行LNA修饰,即锁核酸桥联修饰。LNA锁核酸是指核苷酸糖环上的2'-O与4'-C由亚甲基桥相连的一类RNA衍生物,可以与DNA或RNA按照一般的碱基配对原则进行配对。这种桥连结构可增加核酸骨架的稳定性,提高退火温度,增强碱基配对的特异性,能够极大地降低了错配发生的概率。在本发明中,由于待检测SNPs位点一般有两种基因型,所以本发明优选的同时合成两条LNA修饰的正向引物或两条反向引物;优选的,所述qPCR特异引物与SNPs位点只有单碱基差异,也就是说所述的qPCR特异性标记引物中只有SNPs位点对应的核苷酸有差异,其余核苷酸序列不变。另一条反向引物或正向引物可依据常规引物设计法则进行设计合成,不进行LNA修饰。由于进行LNA修饰的那条引物的位置相对固定,所以在确定是对正向引物还是对反向引物进行LNA修饰时,可参考引物设计法则,选择最容易与DNA链杂交结合的一条引物进行LNA修饰。
在本发明中,设计简并引物进行扩增的目的在于以简并引物扩增获得扩增产物的量与qPCR特异引物扩增获得扩增产物的量相比较来判断qPCR特异引物的扩增是否存在非特异扩增。公知简并引物可以同时扩增SNP位点上两个不同的等位位点,当qPCR特异引物扩增获得扩增产物的量小于等于简并引物扩增获得扩增产物的量时,说明qPCR特异引物的扩增不存在非特异扩增;当qPCR特异引物扩增获得扩增产物的量大于简并引物扩增获得扩增产物的量时,说明qPCR特异引物的扩增存在非特异扩增,后续数据不可靠。
在本发明中,具体的当待测样本为毛果杨时,筛选到的SNP位点优选的位于热激转录因子基因2上SNP8583997位,根据所述SNP位点设计qPCR引物,所述qPCR特异引物包括正向引物SEQ ID NO.1:HSF2SNP8583997T F:5'-ACAGAACTCATGTTCTG-3'、SEQ ID NO.2:HSF2SNP8583997C F:5'-ACAGAACTCATGTTCTG-3'和反向引物SEQ ID NO.4:HSF2R:5'-TAGATGTGCTGCTGACTTCCTAC-3';所述SEQ ID NO.1和SEQ ID NO.2的3'末端上的倒数第二个核苷酸进行LNA修饰。在本发明中,根据所述SNP位点设计的简并引物包括正向简并引物SEQ ID NO.3:HSF2SNP8583997T/C F5'-ACAGAACTCATGTTCTG-3'和反向引物SEQ ID NO.4:HSF2R:5'-TAGATGTGCTGCTGACTTCCTAC-3'。
在本发明中,当待测样品为巨桉时,所述SNP位点位于MYB转录因子基因SNP8583997位上。根据所述SNP位点设计qPCR引物,所述qPCR特异引物包括正向引物SEQ ID NO.5:MYBSNP8583997G F:5'-GAAGACTCTTTGTTTGTCTGTTG-3'、SEQ ID NO.6:MYBSNP8583997A F:5'-GAAGACTCTTTGTTTGTCTGT TG-3'和反向引物SEQ ID NO.8:MYBR:5'-GATCTAGTTGAGTTAGAACAAAG-3';所述SEQ ID NO.5和SEQ ID NO.6的3'末端上的倒数第二个核苷酸进行LNA修饰。在本发明中,设计的简并引物包括正向简并引物SEQ ID NO.7:MYBSNP8583997G/A F:5'-GAAGACTCTTTGTTTGTCTGTTG-3'和反向引物SEQ ID NO.8:MYBR:5'-GATCTAGTTGAGTTAGAACAAAG-3'。
本发明在得到SNPs位点的qPCR特异引物和简并引物后,用所述qPCR特异引物和简并引物分别对待测样本的cDNA进行PCR扩增,获得扩增产物。在本发明中所述的待测样品cDNA是通过提取待测样品的RNA后,进行反转录获得的。本发明对所述的RNA提取和反转录获得cDNA的方法没有特殊限定,采用本领域常规的方法即可。优选的在本发明中,所述待测样本cDNA的PCR扩增程序独立地为:预变性94℃3min;变性94℃10s,退火62℃30s,延伸72℃40s;40个循环。优选的,所述待测样本cDNA的PCR扩增体系独立地包括:20ng/μL的待测样品cDNA,1μL;SYBR Premix Ex TaqTM(2×),10μL;10μM的正向引物0.5μL;10μM的反向引物0.5μL;双蒸水8μL。
本发明在72℃~95℃区间获得PCR产物的熔解曲线。在本发明中,若扩增产物的熔解曲线为单峰,则表明上述的PCR扩增为特异性扩增,然后根据PCR扩增的Ct值计算待测样本等位基因表达特异性。具体的在本发明中,所述待测样本等位基因不平衡表达特异性的计算方法采用2-ΔCt计算方法,ΔCt=候选等位基因基因型R的Ct值-候选等位基因基因型R’的Ct值;所述基因型R为A、T、C和G中的一种,所述基因型R’为A、T、C和G中除R以外的三种中的一种;计算结果即为等位基因不同基因型的不平衡表达差异水平。
下面结合实施例对本发明提供的植物等位基因不平衡表达检测方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
根据Phytozome数据库(https://phytozome.jgi.doe.gov/pz/portal.html#!
gene?search=1&crown=1&detail=1&method=0&searchText=transcriptid:27040781)毛果杨热击转录因子基因2(HSF2)重测序数据筛选位于该基因3’端不保守区域的SNPs位点用于标识等位基因,该SNPs频率在群体中的频率需要大于5%,并且在该位点在实验个体内呈杂合状态。SNP8583997位点满足以上要求,选择该位点为杂合状态的个体作为等位基因表达检测材料。使用LNA修饰引物qPCR检测热激转录因子基因2(Potri.001G108100)的等位基因表达模式,利用毛果杨热击转录因子基因2(HSF2)中SNP8583997作为等位基因标识位点,其所在区域的基因序列如下:
ACTCCAAGTATAGACGTGCTTCAGAACATGAGTTCTGTTACCACTTGCGATTGCTATT,斜体为SNP位点。
针对SNP8583997位点(黑体下划线标注),实验材料体内包含两种类型的模板链,其中模板链I:5'-NNNNNNNNCAGAACATGAGTTCTGTNNNNN
NNNN-3',模板链II:5'-5'-NNNNNNNNCAGAACATGAGTTCTGTNNNNNNN
N-3'。对应的基因型分别为T和C。
设计合成两条3'末端核苷酸LNA修饰(3'末端倒数第二个核苷酸)的正向引物:SEQ ID NO.1(HSF2SNP8583997T F:5'-GGTAACAGAACTCATGTTCTG-3'),SEQ ID NO.2(HSF2SNP8583997C F:5'-GGTAACAGAACTCATGTTCTGG-3'),简并引物SEQ ID NO.3
(HSF2SNP8583997T/C F5'-GGTAACAGAACTCATGTTCTG-3'),反向引物SEQ ID NO.4(HSF2R:5'-TAGATGTGCTGCTGACTTCCTAC-3')。扩增产物目的片段长度为232bp。
用上述引物对高温胁迫处理的毛果杨叶片cDNA进行qPCR扩增。其反应体系为高温(42℃,6h)处理后提取的毛果杨cDNA(20ng/μL),1μL;SYBR Premix Ex TaqTM(2×),10μL;正向引物(10μM),0.5μL;反向引物(10μM),0.5μL;补充双蒸水至20μL。PCR扩增的条件为:预变性94℃3min,变性94℃10s,退火63℃30s,延伸72℃40s,40个循环读板,在72℃-95℃区间获得PCR产物熔解曲线。
PCR扩增曲线如图1所示,其中A为HSF2基因简并引物扩增曲线,B为HSF2SNP8583997T引物qPCR扩增曲线,C为HSF2SNP8583997C引物qPCR扩增曲线。
三个扩增反应的熔解曲线都只出现一个峰,表明都进行了特异性扩增。SEQ ID NO.1与SEQ ID NO.2引物扩增Ct值大于SEQ ID NO.3引物扩增Ct值,表明在等位位点扩增反应中只扩增了引物所对应的等位基因模板。扩增效率在90%-100%之间,表明三对引物扩增效率一致性强,扩增结果可以用于比较分析。
利用即2-ΔCt法计算毛果杨热击转录因子基因2(HSF2)等位基因表达模式,其中ΔCt=(Ct(HSF2-SNP8583997T)-Ct(HSF2-SNP8583997C))。
2-ΔCt=2-(26.81-30.42)
=2-(-3.61)
=12.21
即等位基因HSF2-SNP8583997T表达量比等位基因HSF2-SNP8583997C高12.21倍。
使用ACTIN2作为内参基因,利用即2-ΔΔCt法计算毛果杨热击转录因子基因2(HSF2)等位基因响应高温处理模式。ΔΔCt=(Ct(高温处理-HSF2-SNP8583997T)-Ct(高温处理-ACTIN2))-Ct(对照-HSF2-SNP8583997T)-Ct(对照ACTIN2))。
2-ΔΔCt=2-(26.81-23.56)-(31.12-23.45)
=2-(3.25-7.67)
=2-(-4.42)
=21.4
即等位基因HSF2-SNP8583997T表达量在高温处理下比对照上调21.4倍。
使用ACTIN2作为内参基因,利用即2-ΔΔCt法计算毛果杨热击转录因子基因2(HSF2)等位基因响应高温处理模式。ΔΔCt=(Ct(高温处理-HSF2-SNP8583997C)-Ct(高温处理-ACTIN2))-Ct(对照-HSF2-SNP8583997C)-Ct(对照ACTIN2)))。
2-ΔΔCt=2-(30.42-23.56)-(31.54-23.45)
=2-(6.86-8.09)
=2-(-1.23)
=2.34
即等位基因HSF2-SNP8583997C表达量在高温处理下比对照上调2.34倍。
实施例2
根据Phytozome数据库(https://phytozome.jgi.doe.gov/pz/portal.html#!gene?search=1&crown=1&detail=1&method=0&searchText=transcri ptid:32066262)巨桉MYB转录因子基因(Eucgr.K02470)重测序数据筛选位于该基因3’端不保守区域的SNPs位点用于标识等位基因,该SNPs频率在群体中的频率需要大于5%,并且在该位点在实验个体内呈杂合状态。SNP8583997位点满足以上要求,选择该位点为杂合状态的个体作为等位基因表达检测材料。使用LNA修饰引物qPCR检测MYB转录因子基因(Eucgr.K02470)的等位基因表达模式,利用巨桉MYB转录因子基因(Eucgr.K02470)中SNP 32768853作为等位基因标识位点,其所在区域的基因序列如下:CTCTTTGTTTGTCTGTTGTGTTTATCG,斜体为SNP位点。
针对SNP 32768853位点(黑体下划线标注),实验材料体内包含两种类型的模板链,其中模板链I:5'-NNNNNNNNTGTCTGTTGTGTTTNNNNN
NNNN-3',模板链II:5'-5'-NNNNNNNNGTCTGTTGTGTTTNNNNNNN
N-3'。对应的基因型分别为G和A。进行退火温度梯度PCR扩增,以确定等位基因表达检测的最佳qPCR扩增条件。
设计合成两条3'末端核苷酸LNA修饰(3'末端倒数第二个核苷酸)的正向引物:SEQ ID NO.5(MYBSNP8583997G F:5'-GAAGACTCTTTGTTTGTCTGTTG-3'),SEQ ID NO.6(MYBSNP8583997A F:5'-GAAGACTCTTTGTTTGTCTGT TG-3'),一组简并引物SEQ ID NO.7(MYBSNP8583997G/A F:5'-GAAGACTCTTTGTTTGTCTGTTG-3'),反向引物SEQ ID NO.8(MYBR:5'-GATCTAGTTGAGTTAGAACAAAG-3')。扩增产物目的片段长度为176bp。
用上述对高温胁迫处理的巨桉叶片cDNA进行qPCR扩增。其反应体系为巨桉cDNA(20ng/μL),μL;SYBR Premix Ex TaqTM(2×),10μL;正向引物(10μM),0.5μL;反向引物(10μM),0.5μL;补充双蒸水至20μL。PCR扩增的条件为:预变性94℃3min,变性94℃10s,退火60℃30s,延伸72℃40s,40个循环读板,在72℃-95℃区间获得PCR产物熔解曲线。
PCR扩增曲线如图1所示,其中A为MYB基因简并引物扩增曲线,B为MYBSNP8583997G引物qPCR扩增曲线,C为MYBSNP8583997A引物qPCR扩增曲线。
三个扩增反应的熔解曲线都只出现一个峰,表明都进行了特异性扩增。SEQ ID NO:1与SEQ ID NO:2引物扩增Ct值大于SEQ ID NO:3引物扩增Ct值,表明在等位位点扩增反应中只扩增了引物所对应的等位基因模板。扩增效率在90%-110%之间,表明三对引物扩增效率一致性强,扩增结果可以用于比较分析。
利用即2-ΔCt法计算巨桉MYB转录因子(Eucgr.K02470)等位基因表达模式,其中ΔΔCt=(Ct(MYB-MYBSNP8583997G)-Ct(MYBSNP8583997A))。
2-ΔCt=2-(25.97-28.85)
=2-(-2.88)
=7.36
即等位基因MYBSNP8583997G表达量比等位基因MYBSNP8583997A高7.36倍。
由以上实施例可知,本发明所提供的方法,采用LNA锁核酸修饰的引物对SNP位点进行qPCR扩增以计算等位基因的特异性表达水平,根据实施例中扩增曲线和Ct值可知,所述的特异性强,检测结果的精确性和可靠性,并且仅仅使用PCR手段,简化了实验配套条件,显著地降低了实验成本。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 油菜BnaSPT1基因在促进双子叶植物角果生长中的应用的制造方法与工艺
- 油菜BnaSPT3基因在促进双子叶植物角果生长中的应用的制造方法与工艺
- 一种木质素合成途径中上位性效应检测方法与制造工艺
- 用于检测人类HLA‑B*5701等位基因的核酸、试剂盒及方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- 与癌症相关的基因融合体和基因变异体的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 一种高通量测序检测无创亲子鉴定的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 通过分析预示性基因集诊断亚临床和临床的急性排异的方法与制造工艺