一种增强荧光探针及其制备方法与流程
本发明属于荧光探针领域,涉及一种增强荧光探针及其制备方法,可用于检测单线态氧。
背景技术:
单线态氧,与通常呼吸的稳定基态氧分子的氧气不同,是一种处于激发态氧分子的存在形式。与基态相比,单线态氧的化学性质更活泼,更不稳定,氧化性很强,可以导致各种物质的氧化。单线态氧能够参与多种机体生化反应和生理过程,例如信号传导、酶反应、细胞分裂、组织过氧化、炎症、衰老、吞噬杀菌、肿瘤和化学中毒,对机体产生多种复杂的双重作用。一方面,单线态氧具有较强的毒性,可造成机体的损伤,引发衰老和各种疾病;而另一方面,它又是生物体免疫系统的主要杀菌剂,还可用于癌症的光动力疗法。
目前,在生理条件下的水溶液中定量检测少量单线态氧存在困难,主要在于其寿命短、反应活性高。现有的单线态氧检测的方法主要分为气相中检测方法(例如顺磁共振技术、发射光谱、光电离技术和热量测定等)和液相中检测方法(例如磷光检测法、化学捕获吸光光度法、化学发光法、荧光检测法等)。上述检测方法或成本高难以应用,或者因为单线态氧发光效率低而导致灵敏度低,检出信号弱。
申请号为201610093502.2的中国发明专利公开了一种用于检测单线态氧的染料和荧光探针及其制备方法,通过采用哌嗪衍生物为原料合成对应的荧光探针,并制作成检测试剂盒。该申请中制得的荧光探针,对单线态氧具有高灵敏度和选择性,大大扩展了此类荧光探针的应用范围,例如可用于环境、化学食品和生物医疗领域中的单线态氧检测;但是,该荧光探针的荧光强度仍有提升的空间。
技术实现要素:
本发明目的是为了克服现有技术的不足而提供一种增强荧光探针。
为达到上述目的,本发明采用的技术方案是:一种增强荧光探针,它的化学结构通式如下:
式中,r1和r2相互独立地选自碳原子数为1~6的烷基,z为分子量为500~10000的peg或peg衍生物基团。
优化地,r1和r2均为乙基。
优化地,z为
本发明的又一目的在于提供一种上述增强荧光探针的制备方法,它包括以下步骤:
向反应容器中加入
优化地,所述
由于上述技术方案运用,本发明与现有技术相比具有下列优点:本发明增强荧光探针,通过采用特殊的分子设计使得荧光探针中间体连接上两个荧光基团,而该荧光探针中间体上采用peg或peg衍生物,这样能够极大地提高荧光探针的水溶性,荧光强度得以成倍增强,从而提高了其对特定分子(如单线态氧)的检测效果。
附图说明
附图1为实施例1中增强荧光探针的合成线路图;
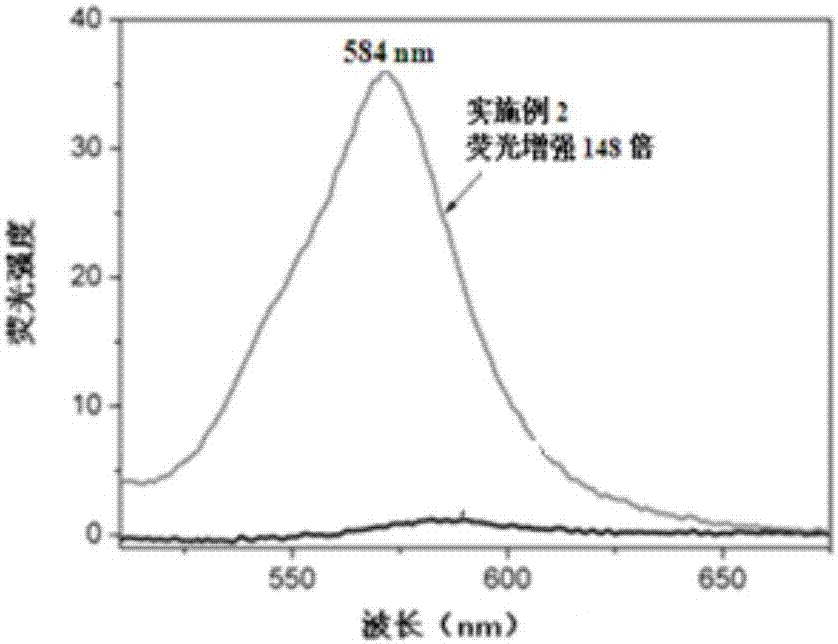
附图2为本发明实施例1中合成的增强荧光探针在室温dmf中与单线态氧反应前后的荧光发射图谱对比(λex=400nm),其中1o2由光激发光敏剂(玫瑰红)15分钟产生;
附图3为本发明实施例1中合成的增强荧光探针在室温水中与单线态氧反应前后的荧光发射图谱对比(λex=400nm),其中1o2由光激发光敏剂(玫瑰红)15分钟产生。
具体实施方式
本发明增强荧光探针,它的化学结构通式如下:
式中,r1和r2相互独立地选自碳原子数为1~6的烷基,z为分子量为500~10000的peg或peg衍生物基团。通过采用特殊的分子设计使得荧光探针中间体连接上两个荧光基团,而该荧光探针中间体上采用peg或peg衍生物,这样能够极大地提高荧光探针的水溶性,荧光强度得以成倍增强,从而提高了其对特定分子(如单线态氧)的检测效果。
r1和r2均优选为乙基。z优选为甲氧基修饰的peg或peg衍生物,r为o、s或nh。上述增强荧光探针的化学结构通式最优为
上述增强荧光探针的制备方法,它包括以下步骤:
向反应容器中加入
而所述
下面将结合附图实施例对本发明进行进一步说明。
实施例1
本实施例提供一种增强荧光探针及其制备方法,该增强荧光探针的化学结构式如下:
(a1)在50ml三口瓶中,加入hocd(1g,2.43mmol,1eq,即
(a2)在50ml单口瓶中,加入第一产物(800mg,1.38mmol,1eq),加入氯磺酸(16ml),加热回流24小时,将反应液加到20ml冰水中,用二氯甲烷提取,浓缩过柱纯化得到化合物2652mg(71%)蓝色固体(第二产物
(a3)在50ml单口瓶中,加入第二产物(200mg,0.3mmol,1eq),加入二氯甲烷(5ml),et3n(三乙胺,37mg,0.36mmol,1.2eq),甲氧基聚乙二醇(即mpeg-oh,mw=1000)(23mg,0.36mmol,1.2eq),50℃下反应5小时,冷却到室温,加冰水1ml,浓缩,过柱纯化得到第三产物144mg(即
(b1)取哌嗪(5.31mg,0.062mmol)溶于干燥二氯甲烷(dcm)(6ml)中,然后加入苯并三氮唑-n,n,n',n'-15四甲基脲六氟磷酸盐(hbtu)(34.3mg,0.091mmol)、n,n-二异丙基乙基胺(dipea)(31μl,0.184mmol)后,与
(c)在50ml单口瓶中,加入第三产物(100mg,0.141mmol,1eq)、ch3cn(5ml),k2co3(98mg,0.705mmol,5.0eq)和
实施例2
本实施例提供一种增强荧光探针及其制备方法,该增强荧光探针的化学结构式和反应步骤与实施例1中的基本一致,不同的是:(a3)中使用的mpeg-oh分子量为10000,步骤(b1)中使用的原料为
实施例3
本实施例提供一种增强荧光探针及其制备方法,该增强荧光探针的化学结构式和反应步骤与实施例1中的基本一致,不同的是:(a3)中使用的mpeg-oh分子量为500。
取3份光敏剂玫瑰红(2μl,0.5mg/ml,溶于10mmpbs缓冲液,ph=7.4)储藏液,分别加入实施例1至实施例3中的探针混合;将溶液暴露在纤维照明器的光线下数分钟,并用荧光计记录荧光强度变化。其中,实施例1中的实验结果图2所示,增强荧光探针在dmf(1μm)与单线态氧反应后荧光激发谱强度分别增强了148倍;而实施例2和实施例3中制得的增强荧光探针在dmf(1μm)与单线态氧反应后荧光激发谱强度分别增强了146倍、145倍(以申请号为201610093502.2中国发明专利中的iiia探针作为对比例,测得增加了60倍),显然它们大大高于对比例。在模拟生物生理环境的pbs缓冲液中,对比例1中浓度为0.1μm的探针与单线态氧反应后荧光激发谱分别强度了120倍(如图3所示);而实施例2和实施例3中制得的增强荧光探针在pbs缓冲液中与单线态氧反应后荧光激发谱强度分别增强了116倍、115倍,约为在有机溶剂dmf中测到的80%,这说明该探针的水溶性并未比油溶性降低多少,具有良好的水溶性。
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围,凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!