苯并二氮杂*类化合物的制作方法
本发明涉及一种短3-苯并二氮杂
背景技术:
苯并二氮杂草类药物在临床上广泛用于抗焦虑、镇静和催眠。作为第一个水溶性苯并二氮杂草类衍生物,咪达唑仑已经广泛用于临床镇静、催眠、镇痛、抗癫痫、抗焦虑和全身麻醉。当咪达唑仑作为麻醉药输入人体后,可被细胞色素p450同工酶氧化为α-羟基咪达唑仑。但该氧化物仍具有药理活性,因而麻醉作用时间长,苏醒慢。因此研发麻醉诱导时间快、维持时间短的新型苯并二氮杂草类水溶性衍生物一直为药物化学家所重视。
式(ii)化合物的化学名为3-[(4s)-8-溴-1-甲基-6-(2-吡啶基)-4h-咪唑[1,2-a][1,4]苯并二氮杂卓-4-基]丙酸甲酯,
其中,r为氢、甲基、乙基、异丙基。
专利ep1183243中报道该类化合物是短效中枢神经系统(cns,centralnervoussystem)抑制剂,具有包括镇静催眠、抗焦虑、肌肉松弛和抗惊厥作用。可用于以下临床治疗方案中的静脉给药:如手术期间中的手术前镇静、抗焦虑和遗忘用途;在短期诊断、手术或内窥镜程序期间的清醒性镇静;在施用其它麻醉剂和止痛剂之前和/或同时,作为用于全身麻醉的诱导和维持的组分;icu镇静等。该化合物代谢迅速,不依赖于细胞p450酶代谢,可通过多种器官代谢,且其代谢产物活性很低,减少了药物之间的相互作用,同时也为代谢器官功能受损患者的使用提供了可能。
但是,式(ii)所示化合物极不稳定,在强制降解的影响因素试验中,很容易产生降解杂质,这些降解杂质有的含量很少,需要长时间的分离富集进行结构鉴定,至今为止,尚未见到有关这些降解杂质的分离、结构确认及其用途研究的相关报道。
技术实现要素:
我们在对式(ii)进行稳定性研究时,发现该化合物很不稳定,可产生较多的降解杂质。因此,将式(ii)所示化合物与苯磺酸或对甲苯磺酸成盐以增加其稳定性,发现其苯磺酸盐或对甲苯磺酸盐比其碱基的稳定性大大增强,但在强制降解的稳定性研究中仍会产生降解杂质,但这些杂质在此之前均未发现有文献报道过。我们花费一年多的时间,通过制备型液相将这些杂质进行了分离,并进行了结构确认,发现其中有一个降解杂质的结构式如式(i)所示结构:
其中,r为氢、甲基、乙基、异丙基。
随后,我们对该杂质的结构进行了合成研究。
因此,本发明的目的在于提供一种式(ii)所示化合物的降解杂质,具有式(i)所示结构。本发明的另一目的在于提供一种式(i)所示结构的合成方法,其特征在于式(ii)所示化合物与碱进行水解反应下得到;
进一步,其中所述水解反应温度为-40~0℃。
本发明的另一目的还在于提供一种式(i)所示结构的合成方法,将式(ii)所示化合物先与碱进行水解反应、然后再进行溴代反应得到;
进一步,式(ii)所示化合物与碱发生水解反应的温度为~100℃;
进一步,所述的溴代反应的溴代试剂为溴化氢、乙酰溴、溴素、nbs;
进一步,所述的溴代反应的温度为-10~110℃;
进一步,所述的溴代反应是在非质子性溶剂中发生的;
进一步,所述的溴代反应的非质子溶剂选自四氢呋喃、乙腈、dmf、丙酮、二氯甲烷、三氯甲烷、二氯乙烷、甲苯、二甲苯任一种或其组合。
进一步,上述两种方法中,所述的水解反应是在质子性溶剂中发生的;
进一步,所述的水解反应的质子性溶剂为甲醇、乙醇、异丙醇、丁醇、水任一种或其组合;
进一步,所述的碱为氢氧化钠、氢氧化钾、氢氧化锂、氢氧化钙、氢氧化钡、碳酸钠、碳酸钾的任一种或其组合。
进一步,本发明式(i)所示化合物的分离、纯化方法选自柱层析、重结晶或制备型液相分离纯化中的任一种或其组合。
进一步,本发明所述的重结晶方法为式(i)所示化合物的有机酸盐在甲醇、乙醇、丙酮、2-丁酮的任一种或其组合中进行。
进一步,本发明所述的式(i)所示化合物的有机酸盐是指盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、草酸盐、马来酸盐、对甲苯磺酸盐或甲苯磺酸盐。
进一步,本发明所述的柱层析分离精制的条件为,洗脱剂为乙酸乙酯-石油醚、乙酸乙酯-正己烷的任一种或其组合,进行等度或梯度洗脱,收集产品流份,浓缩或冻干干燥,即得。
进一步,本发明所述的制备型液相分离精制的条件为,流动相为甲醇-乙腈、甲醇-乙腈-水、甲醇-水的任一种或其组合,进行等度或梯度洗脱,收集产品流份,浓缩或冻干干燥,即得。
本发明的式(i)所示化合物用作标准品或对照品在药物分析的应用。
发明人通过对式(i)所示化合物的研究发现,本发明的式(i)所示化合物具有较好的抗血栓活性,并对其毒性进行了初步的研究。因此,本发明的另一目的在于提供式(i)所述的化合物或其组合物在抗血栓药物中的应用。血栓性疾病是心肌梗死、缺血性心肌病、脑血栓、脑栓塞、脑梗塞、短暂性脑缺血发作或腔隙性脑梗塞。本发明的另一目的在于提供一种新型的抗血栓组合物,组合物中含有有效剂量的式(i)所示的化合物,或其药学上可接受的盐、立体异构体、互变异构体。
本发明中式(i)化合物的药学上可接受的盐是指盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、草酸盐、马来酸盐、对甲苯磺酸盐或甲苯磺酸盐。
本发明中式(i)化合物通过药物制剂、医疗器械、日化用品形式得以体现;所述药物制剂、医疗器械、日化产品的形式是水凝胶、泡沫凝胶、涂膜剂、巴布剂、冻干粉、水剂、气雾剂、栓剂、外用搽剂、软膏剂。
本发明中式(i)化合物的药物制剂的药物赋形剂,包括传统的抗粘剂、粘合剂、崩解剂、填料、稀释剂、助流剂、润滑剂和防腐剂。
粘合剂包括但不限制于明胶、纤维素、改性纤维素(如微晶纤维素)、甲基纤维素、聚乙烯吡咯啶酮、淀粉、蔗糖和聚乙二醇;特别优选聚乙烯吡咯啶酮和/或微晶纤维素。在一个优选实施方案中,抗粘剂的含量在从0.001至30重量%,更优选0.1至25重量%的范围内。
填料和/或稀释剂,优选选自但不限制于由纤维素、二磷酸钙、乳糖、蔗糖、葡萄糖、甘露醇、山梨醇和碳酸钙所组成的组;特别优选微晶纤维素和乳糖。在一个优选实施方案中,填料和/或稀释剂的含量在从0.001至90重量%,更优选0.1至80重量%,最优选10至75重量%的范围内。
润滑剂,如硬脂酸镁、硬脂酸和硬酯。在一个优选实施方案中,润滑剂的含量在从0.001至5.0重量%的范围内。
崩解剂,如交联羧甲基纤维素钠(交联羧甲纤维素钠)、交联聚乙烯吡咯啶酮和淀粉乙醇酸钠。在一个优选实施方案中,崩解剂的含量在从0.001至5.0重量%的范围内。
抗氧化剂,如维生素a、维生素e、维生素c、棕榈酸视黄酯和硒;半胱氨酸、甲硫氨酸、柠檬酸、柠檬酸钠、苯甲酸甲酯和苯甲酸丙酯。
附图说明
图1式(i)所示化合物(r为甲基)的hplc图,
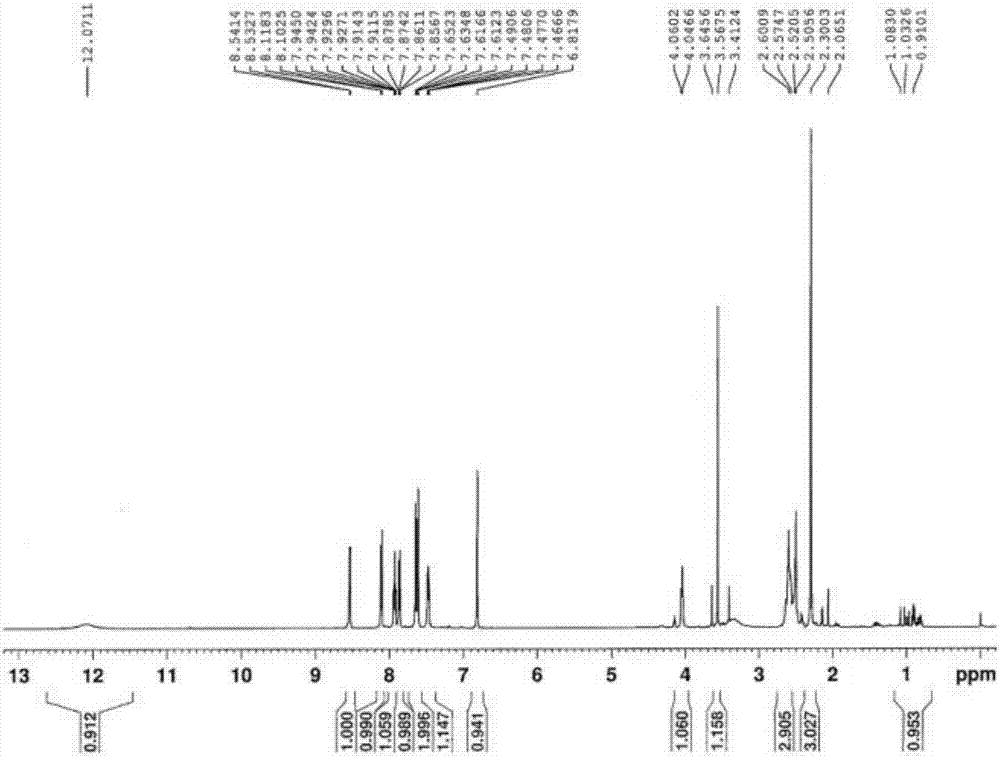
图2式(i)所示化合物(r为甲基)的1h-nmr图
图3式(i)所示化合物(r为甲基)的质谱图([m-h]+)
图4式(i)所示化合物(r为甲基)的质谱图([m+h]+)
图5式(i)所示化合物(r为氢)的质谱图([m+h]+)
图6式(i)所示化合物(r为乙基)的质谱图([m-h]+)
图7式(i)所示化合物(r为异丙基)的质谱图([m+h]+)
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。实施例中未注明的具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。
在本发明的实施例中,
hplc:分析柱:diamonsiltmc18(250mm×4.6mm);流动相:0.01mol·l-1磷酸二氢钾(0.3mmol·l-1的十二烷基磺酸钠)-乙腈(65:35);流速:1ml·min-1;紫外检测波长:240nm;进样量:10μl。
1h-nmr:avanceiii500m全数字化超导核磁共振谱仪
质谱:brukerapexiv傅立叶变换回旋共振质谱仪。
实施例1:式(i)所示化合物(r为甲基)的制备
将5g式(ii)所示化合物(r为甲基)溶于50ml甲醇中,降温至-40℃,滴加10%的氢氧化钠水溶液调ph至9~10,并始终使反应液保持该ph值,保持反应温度-40~-30℃搅拌至反应完成,用稀盐酸水溶液洗涤ph至6~7,用200ml二氯甲烷萃取,二氯甲烷层用无水硫酸镁干燥过夜,浓缩,过柱(洗脱剂为乙酸乙酯:石油醚=1:15),洗脱液减压浓缩,残留物用少量乙醇加热至60℃,加入1g草酸,搅拌30min,降温至0~5℃搅拌析晶,过滤,滤饼用碳酸氢钠中和,干燥,得式(i)所示化合物0.96g,hplc面积归一化法,测得其含量为99.7%。
mp:210.2℃~211.9℃
ms:425.0[m+h]+,423.1[m-h]+
1h-nmr(500mhz,dmso-d6)
δ:0.91~1.08(m,1h),2.30(s,3h),2.50~2.60(m,3h),3.56(s,1h),4.04~4.06(d,1h),6.81(d,1h),7.47~7.49(m,1h),7.61~7.65(m,2h),7.85~7.87(m,1h),7.91~7.95(m,1h),8.10~8.11(m,1h),8.53~8.54(m,1h)12.07(s,1h)。
实施例2:式(i)所示化合物(r为氢)的制备
将5g式(ii)所示化合物(r为氢)溶于35ml乙醇中,降温至-30~-20℃,滴加5%氢氧化钾水溶液调ph至10~12,在反应过程中保持该ph值,-20~-10℃保温反应,反应完毕加入200ml二氯甲烷,用乙酸水溶液洗涤ph至6~7,无水硫酸镁干燥过夜,浓缩,过柱(洗脱剂为乙酸乙酯:正己烷=1:20),洗脱液减压浓缩,残留物用少量丙酮加热至45℃,加入1.6g马来酸,搅拌30min,降温至0~5℃搅拌析晶,过滤,滤饼用碳酸氢钠中和,干燥,得式(i)所示化合物1.57g。
mp:203.4℃~205.1℃
ms:411.2[m+h]+
实施例3:式(i)所示化合物(r为乙基)的制备
将5g式(ii)所示化合物(r为乙基)溶于60ml异丙醇中,降温至-10~-5℃,分批加入一水合氢氧化锂0.5g,室温搅拌至反应完成,加入200ml二氯甲烷,用稀盐酸溶液洗涤ph至6~7,无水硫酸镁干燥过夜,浓缩,过柱(洗脱剂为乙酸乙酯:石油醚=1:10),洗脱液减压浓缩,残留物用制备型液相进行分离,流动相为甲醇:乙腈:水:三乙胺=45:25:30:0.01,接收馏分并浓缩,得式(i)所示化合物1.15g。
mp:213.4℃~215.1℃
ms:437.1[m-h]+
实施例4:式(i)所示化合物(r为异丙基)的制备
将5g式(ii)所示化合物(r为异丙基)溶于30ml正丁醇和10ml水中降至0℃,分批加入碳酸钾1.58g,室温搅拌至反应完成,加入200ml二氯甲烷,用盐酸水溶液洗涤ph至6~7,无水硫酸镁干燥过夜,浓缩,过柱(洗脱剂为乙酸乙酯:环己烷=1:15),洗脱液减压浓缩,残留物用少量甲醇加热至50℃,加入1.6g富马酸,搅拌30min,降温至0~5℃搅拌析晶,过滤,滤饼用碳酸氢钠中和,得式(i)所示化合物1.48g。
mp:214.6℃~216.4℃
ms:453.4[m+h]+
实施例5:式(i)所示化合物(r为异丙基)的制备
将5g式(ii)所示化合物(r为异丙基)溶于15ml甲醇和10ml水中,室温加入氢氧化钠0.45g,45℃搅拌至反应完成,加入300ml二氯甲烷,用盐酸水溶液洗涤ph至6~7,无水硫酸镁干燥过夜,浓缩,残留物用50ml甲苯溶解,加热至110℃,滴加40%的溴化氢乙酸溶液8ml,在110~120℃反应至完毕,60℃减压浓缩出溶剂,残留物用100ml二氯甲烷溶解,用饱和碳酸氢钠溶液中和至中性,二氯甲烷层无水硫酸钠干燥,过滤,浓缩,残留物过柱(洗脱剂为乙酸乙酯:石油醚=1:30),洗脱液减压浓缩,残留物用少量2-丁酮加热至45℃,加入2g酒石酸,搅拌30min,降温至0~5℃搅拌析晶,过滤,滤饼用碳酸氢钠中和,干燥,得式(i)所示化合物0.36g。
实施例6:式(ii)所示化合物(r为乙基)的制备
将5g式(ii)所示化合物(r为乙基)溶于25ml乙醇和5ml水中,室温加入碳酸钠1.5g,65℃搅拌至反应完成,加入300ml二氯甲烷,用盐酸水溶液洗涤ph至6~7,无水硫酸镁干燥过夜,浓缩,残留物用50ml二甲苯溶解,加热至110℃,滴加乙酰溴10ml,在110~120℃反应至完毕,80℃减压浓缩出溶剂,残留物用100ml二氯甲烷溶解,用饱和碳酸氢钠溶液中和至中性,二氯甲烷层无水硫酸钠干燥,过滤,浓缩,残留物用制备型液相进行分离,流动相为甲醇:乙腈:水:三乙胺=45:25:30:0.01,接收馏分并浓缩,,得式(i)所示化合物0.47g。
实施例7:式(i)所示化合物(r为甲基)对血小板聚集活性的药理实验方法与结果
实验方法:取家兔2只,用利多卡因局部麻醉,手术分离颈总动脉取血,采取3.8%枸橼酸钠1:9抗凝,以500r/min离心10min,制备富血小板血浆(prp),剩余部分再以3000r/min离心,制备贫血小板血浆(ppp),按比浊法进行血小板聚集实验。测定管中加入prp240μl、不同浓度受试药物30μl,37℃温孵5min,分别以30μl二磷酸腺苷钠盐(adp)(终浓度为10μmol/l)为诱导剂,观察记录5min内最大聚集率。以dmso作空白对照阿司匹林作阳性对照,计算目标化合物的血小板聚集抑制率(air)。
测试结果:表1中列出了本发明式(i)化合物对adp诱导家兔血小板聚集活性数据,阳性对照药为阿司匹林
表1本发明部分化合物对adp诱导的家兔血小板聚集的抑制活性
以上药理学数据显示,本发明式(i)化合物能够抑制血小板聚集,且具有比阿司匹林更强的血小板聚集抑制作用。
r为氢、乙基、异丙基时也得到类似结果。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于二溴1,4?二丙基?1,4?二氮杂二环[2.2.2]辛烷的二价锰荧光材料及其制备方法 ...的制作方法
- 一种苯并二氮杂卓类化合物作为农用杀虫剂的新用图
- 一种苯并二氮杂卓类化合物作为杀菌剂的用图
- 一种(1s,4s)?2,5?二氮杂二环[2.2.2]辛烷及其衍生物的合成方法
- 包含噻吩并三唑并二氮杂卓化合物的药物制剂的制作方法
- 一种苯并内酯类化合物、其制备方法和在制备抗癌药物中的应用
- 二氮杂-苯并荧蒽并烯类化合物的制作方法
- 制备(2s,5r)-1,6-二氮杂-二环[3.2.1]辛烷-2-腈-7-氧代-6-(磺氧基)-单钠盐的方法
- 利用含有噻吩并三唑并二氮杂*化合物的药物制剂治疗白血病的方法
- 一种注射用短效苯并二氮杂卓盐药物组合物及其制备方法
- 一种制备(1s,4s)?2,5?二氮杂二环[2.2.1]庚烷或[2.2.2]辛烷衍生物的方法
- 一种基于二溴1,4?二丙基?1,4?二氮杂二环[2.2.2]辛烷的二价锰荧光材料及其制备方法 ...的制作方法
- 一种苯并二氮杂卓类化合物作为农用杀虫剂的新用图
- 一种苯并二氮杂卓类化合物作为杀菌剂的用图
- 一种(1s,4s)?2,5?二氮杂二环[2.2.2]辛烷及其衍生物的合成方法
- 包含噻吩并三唑并二氮杂卓化合物的药物制剂的制作方法
- 一种苯并内酯类化合物、其制备方法和在制备抗癌药物中的应用
- 二氮杂-苯并荧蒽并烯类化合物的制作方法
- 制备(2s,5r)-1,6-二氮杂-二环[3.2.1]辛烷-2-腈-7-氧代-6-(磺氧基)-单钠盐的方法
- 利用含有噻吩并三唑并二氮杂*化合物的药物制剂治疗白血病的方法
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- 一种苯并咪唑类化合物的合成方法
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗缺氧药物的制作方法
- 一种含苯并烯氟菌唑和氟嘧菌酯的杀菌组合物的制作方法
- 一种苯并二氮杂卓类化合物作为杀菌剂的用图
- 一种氟唑菌苯胺和丁香菌酯的杀菌组合物的制作方法