亲水性药物的靶向递送的制作方法
发明领域本发明涉及将亲水性药物给药到人体或动物的肝脏,以及给药到实体肝脏瘤。更具体来说,本发明涉及亲水性药物,特别是抗癌药物的给药。
背景技术:
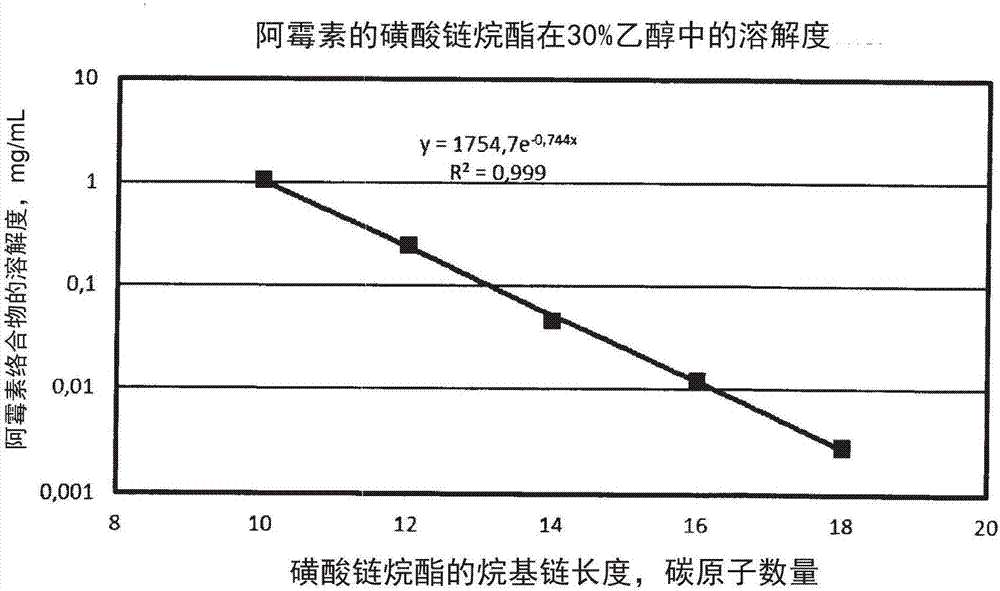
:治疗窗口表示可以对疾病进行有效和安全治疗的药物剂量范围。其包括观察到明显治疗效果的剂量至治疗益处被不利影响中和的剂量。大部分的抗癌药物具有窄治疗窗口。此外,通常很少比例的给药剂量到达待治疗位点。通过口服摄取或血管注射进行系统给药之后,药物经由循环分布在整个人体,导致整个人体受到影响。理想地,药物应该被专门导向到所需的体部位,例如需要进行治疗的器官或组织。该靶向给药会避免对身体的其余部分造成伤害。这种给药类型需求将药物导向到感兴趣的组织,同时避免大量药物抵达不需要治疗的组织。需要被导向到特定身体部位的药物例子是抗癌药物阿霉素。通常接受的是,通过靶向药物递送,可以明显改善阿霉素的治疗潜力,因为由此可以避免或者至少明显降低其有害副作用。阿霉素的最危险副作用是对心脏的损害。当阿霉素的累积剂量达到550mg/m2时,发生心脏副作用的风险急剧增加。阿霉素心脏中毒表征为线粒体氧化磷酸化中的剂量依赖性下降。由于阿霉素与铁相互作用产生的反应性氧物质会损伤肌细胞,导致肌原纤损耗和细胞质空泡形成。此类过度损伤可能导致患者死亡。因此,希望保持阿霉素的心脏浓度尽可能的低。肝癌是需要给药阿霉素的许多恶性肿瘤之一。更具体来说,可以通过称作经动脉化疗栓塞的工艺进行靶向(或局部)给药来治疗肝癌。在该过程中,将阿霉素通过导管直接给药到动脉,给药到肝脏的患病部分,同时通过例如海绵胶堵住未受到疾病影响的肝脏的动脉供给部分。隔离肝脏输注是用于肝脏的药物靶向递送的一种方法。其包括外科手术,在该过程中,从体循环隔离到达肝脏的血液循环,并用血液分开输注。在隔离之后,通过输注到肝脏的血液注入药物,实现较高浓度的化学治疗试剂用于系统给药。但是,该侵入式过程在技术上是复杂且不安全的。许多药理活性剂,例如前述的抗癌药是包含一个或多个氨基的弱碱。由于该原因,它们与强酸和弱酸形成盐,并且通常以盐形式给药。它们的常见的药学可接受盐(特别是它们的盐酸盐、氢溴化物、磷酸盐、硫酸盐、乳酸盐、酒石酸盐等)在水性体流体中的溶解度通常高于游离碱的溶解度。因此,此类盐的水性溶液而不是相应碱的水性溶液被用于静脉注射。为了给药到人体或动物肝脏,此类抗癌药以阳离子两亲形式提供(药学可接受酸的盐形式)。这种给药方式应用于抗癌药,例如,蒽环霉素(阿霉素、表柔比星、柔红霉素、伊达比星、米托蒽醌),长春花属生物碱(长春碱、长春新碱、长春瑞滨),安吖啶,拓扑替康和伊立替康,但不限于此。如果药学活性剂包含不止一个氨基,则它们中的两个或更多个可以质子化并与酸形成盐。前述类型的阳离子两亲药物(cad)与两亲阴离子表面活性剂(例如,磺酸链烷酯)反应,形成不溶于水的络合物。虽然仍然符合有机碱与有机酸或无机酸的盐的定义,但是不溶于水的络合物在一定程度上额外通过非共价键作用力连接。发明目的本发明的一个主要目的是提供用于肝脏抗癌药物的靶向给药的药学组合物,其包含一个或多个氨基官能团,其不含已知药物组合物的一种或多种缺陷或者至少在一定程度上展现出对于这些缺陷的减少。本发明的另一个目的是提供用于实体肝脏瘤抗癌药物的靶向给药的药学组合物,其包含一个或多个氨基官能团,其不含本领域已知药物组合物的一种或多种已知缺陷或者至少在一定程度上展现出对于这些缺陷的减少。本发明的另一个目的是提供一种此类药学组合物的设计方法,其在静脉给药之后,会在肝脏或实体肝脏瘤中提供所需的靶向药物浓度。通过研究本发明的如下简要描述、其优选实施方式以及所附权利要求,本发明的其他目的会是显而易见的。技术实现要素:本发明基于如下理解:两亲化合物(特别是包含氨官能团的亲水性抗癌药物的磺酸直链烷基酯和硫酸直链链烷酯)颗粒的水性悬液是有价值的形式,通过这种形式,可以将这些药物以集中其治疗效果的方式给药到肝脏或实体肝脏瘤,即,肝脏或实体瘤的靶向定位。因此,靶向定位理解为优先递送到肝脏从而在肝脏组织(包括肝癌组织)中实现相比于其他组织更高的药物浓度(w/w)。亲水性抗癌药物的磺酸直链烷基酯和硫酸直链链烷酯的一个重要特征在于它们在水和水性体液中在25℃时小于0.1mg/ml的低溶解度。作为本发明背后的一种可能的生物学解释(但不以任何方式约束本发明)是,在向系统循环或肝脏或实体肝脏瘤给药了此类抗癌药物的颗粒水性悬液之后,药物颗粒会在给定时间段内在体内达到平衡分布。它们在水性介质中的溶解度是非常低的,但并非为零。因此,它们会缓慢溶解在体液中,直至达到由它们的溶解度决定的平衡。由于溶解的材料不可逆地化学转变成降解产物,随时间溶解了更多的材料,从而维持平衡。只要通过材料溶解供给了平衡,就局部地维持了药物的稳态浓度。本发明还基于如下理解:包含氨官能团的亲水性抗癌药物的两亲磺酸直链烷基盐/酯的水性胶体以及包含氨官能团的亲水性抗癌药物的两亲硫酸直链链烷盐/酯的水性胶体是有价值的形式,通过这种形式,可以将这些药物以集中其治疗效果的方式给药到所需器官(特别是肝脏),或者给药到所需实体瘤(即,器官或实体瘤的靶向定位)。水性胶体由粒度最高至约10000nm的颗粒构成,或者包括粒度最高至约10000nm的颗粒。本发明的水性胶体的一个重要性质在于它们的低沉淀速率。通常来说,随着粒度的增加,给定颗粒种类的沉淀速率增加。但是,通过增加水性相的粘度和/或通过改变颗粒的表面性质(例如,表面粗糙度),可以防止其发生增加并且甚至可以使其下降。本发明提供了亲水性癌症药物的盐的固体颗粒,其包含一个或多个氨基以及水溶性硫酸烷基盐/酯或磺酸链烷盐/酯或者两种或更多种此类硫酸盐/酯或磺酸盐/酯的混合物。盐的一个重要特征在于其在水中的低溶解度。换言之,本发明的盐是基本不可溶的。“基本不可溶”应理解为在水中或水性体液中的溶解度小于0.1重量%,特别地,小于0.05或0.02重量%。本发明提供了具有水溶性硫酸烷基酯或磺酸链烷酯或者具有此类硫酸酯或磺酸酯中的两种或更多种的混合物的亲水性癌症药物的不溶于水的盐的固体颗粒的生产方法。本发明还提供了药学组合物,其包含一种或多种本发明的两亲磺酸盐/酯和/或硫酸盐/酯以及液体载剂。可以通过任意合适途径,例如动脉内、腹膜内、肌肉内、透皮或静脉给药方式,来给药该组合物。优选以一剂包含本发明的两亲磺酸盐/酯和硫酸盐/酯的水性胶体的进行给药。本发明还提供了包含亲水性癌症药物的水不溶性盐和水溶性硫酸烷基酯或磺酸链烷酯,或者两种或更多种此类硫酸盐或磺酸盐的混合物的固体颗粒形式的药物组合物的生产方法。本发明的组合物还可包含缓冲剂和药学可接受赋形剂,例如渗透压(osmolality)控制剂和粘度控制剂。由于采用的生产方法,组合物额外含有由水溶性硫酸烷基酯或磺酸链烷酯的阳离子以及抗癌药物的阴离子构成的盐或对应离子。优选使用碱性硫酸烷基酯和碱性磺酸链烷酯,特别是硫酸烷基钠和硫酸烷基钾以及磺酸链烷钠和磺酸链烷钾。本发明的两亲颗粒磺酸盐/酯和硫酸盐/酯由药理学试剂d和一种或多种硫酸酯或磺酸酯阴离子构成,所述药理学试剂d具有抗癌活性,其包含1-4个氨基(其中的一个或多个被质子化)。它们表示为如下化学式(1)和(2):dn+(r1so3)-n(1)dn+(r2oso3)-n(2)其中,r1是直链c6-c30烷基;r2是直链c6-c30烷基;n是1-4的整数。r1和r2优选是直链c10-c20烷基,更优选是直链c12-c18烷基,甚至更优选约为直链c16烷基。因此,r1可以是直链c12、c13、c14、c15、c16、c17、c18烷基中的任意一种;r2是直链c12、c13、c14、c15、c16、c17、c18烷基中的任意一种。90%的胶体颗粒的优选粒度小于或等于10000nm,更优选小于或等于5000nm。根据本发明的一个优选方面,本发明包括尺寸大于胶体颗粒的颗粒以及它们的水性悬液,例如,粒度最高至10μm或50μm以及甚至最高至100μm,以及它们的悬液。可以通过例如,离心或冷沉淀,从水性相分离本发明的颗粒。如果通过离心进行分离,则水性相消除了伴随而来的由水溶性硫酸烷基酯或磺酸链烷酯的阳离子以及抗癌药物的阴离子构成的盐或对应离子。所得到的粉末(如果需要的话,进行额外干燥)保留至少50%,更优选至少80%的胶体粒度。为了促进在水性介质中的再悬浮,粉末可以包含再悬浮促进剂,例如葡萄糖、乳糖或白蛋白。或者,可以通过水性介质的蒸发(包括冷沉淀)生产本发明的颗粒;以这种方式,它们会与伴随而来的盐混合,所述盐包含水溶性硫酸烷基酯或磺酸链烷酯的阳离子和抗癌药物的阴离子;如果需要的话,它们可以与再悬浮促进剂混合。根据本发明的另一个优选方面,本发明的胶体颗粒可以包含对于待治疗的肿瘤的表面抗原具有亲和力的微载体颗粒,例如通过包含合适的抗体结构。本发明的两亲磺酸盐/酯和硫酸盐/酯的优选药理学活性剂d包括但不限于:阿霉素、表柔比星、柔红霉素、伊达比星(idarubicin)、米托蒽醌、长春碱、长春新碱、长春瑞滨、安吖啶、拓扑替康、伊立替康。根据本发明的另一个优选方面,除了上文所述的那些,用于制备本发明的两亲磺酸盐/酯和硫酸盐/酯的合适的抗癌剂是具有与阿霉素、表柔比星、柔红霉素、伊达比星、米托蒽醌、长春碱、长春新碱、长春瑞滨、安吖啶、拓扑替康、伊立替康类似的亲水性。根据本发明,还揭示了治疗人体中的肝癌的方法,其包括:向所述人体给药治疗有效量的本发明的药物组合物或者包含本发明的两亲颗粒磺酸盐/酯或硫酸盐/酯粉末的药物组合物。一种优选的给药方法是通过输注或注射进入静脉或动脉,特别是进入门静脉或肝动脉。另一种优选的给药方法是通过输注或注射进入外周循环到达实体肝脏瘤。第三种优选的给药方法是直接输注或注射进入实体肝脏瘤。根据本发明的一个优选方面,优选通过一剂或者多剂给药。向肝脏或肝脏瘤给药的一个方面是通过输注或注射进入静脉或动脉,特别是进入门静脉或肝动脉。根据本发明,提供了用于方便和非侵入式给药的药物递送系统,其能够在延长的时间段上(例如,超过1小时或6小时或者甚至大于或等于1天)提供所需的药物浓度。在肝脏或者实体肝脏瘤或者其他组织中提供并维持了此类浓度。根据本发明的一个优选方面,本发明提供了控制肝脏与其他器官和组织之间的药物分布比的方法。在本申请中,除非另有说明,否则术语“肝脏疾病”(肝病)包括原发性肝癌(例如,肝细胞癌和/或胆管癌、血管肉瘤、肝的血管肉瘤),继发性恶性肿瘤(即,从原发性癌症转移的胃肠道和其他器官(例如肾、肺、乳腺或前列腺)中的继发性损伤);由于病毒引起的肝脏感染(病毒性肝炎)、肝毒素(例如酒精性肝炎)、自身免疫性(自身免疫性肝炎)或遗传性病症;过度饮酒引起的肝硬化。根据本发明,还揭示了药物组合物的设计方法,其用于在预定时间段内,为选自肝脏或实体肝脏瘤的人或动物的治疗靶标提供预定浓度的药理学活性剂d的硫酸盐/酯或磺酸盐/酯(其包含1-4个如下化学式(1)或(2)表示的氨基)或者这些试剂的混合物:dn+(r1so3)-n(1)dn+(r2oso3)-n(2)其中,r1是直链c6-c30烷基;r2是直链c6-c30烷基;n是1-4的整数;其中,该方法包括:i)对于各种碳链长度x、y,确定dn+(r1so3-)n和/或dn+(r2oso3-)n在水性溶剂中的溶解度;ii)在将所述药理学活性剂d给药到对象或动物之后,确定所述药理学活性剂的所述硫酸盐/酯或磺酸盐/酯的溶解度与所述药理学活性剂d在治疗靶中的预期浓度之间的相关性;iii)基于所述药理学活性物质d在所述器官或组织中的所需浓度,限定所述药理学活性剂的所述硫酸盐/酯或磺酸盐/酯在所述溶剂中的目标溶解度;iv)确定对应所述目标溶解度的碳链长度x、y;v)提供包含由此确定的碳链长度x、y的所述药理学活性剂的硫酸盐/酯或磺酸盐/酯;vi)提供流体载剂;vii)将包含由此确定的碳链长度x、y的所述药理学活性剂的硫酸盐/酯或磺酸盐/酯与流体载剂组合,组合的量能够在所述时间段内维持所述浓度。根据本发明的一个优选方面,确定水性有机溶剂(例如,水性醇,特别是浓度5-50%,优选10-30%(v/v)的水性乙醇)中的溶解度。也可使用其他与水易混溶的溶剂,例如,低分子量酮、酰胺、酯、酰胺和亚砜。根据本发明的一个优选方面,药物组合物包含:化学式(1)或(2)表示的本发明的至少两种不同硫酸盐/酯或磺酸盐/酯的混合物,或者至少两种不同的硫酸盐/酯或磺酸盐/酯(其中一个由化学式(1)表示,另一个由化学式(2)表示)的混合物。优选的药理学活性物质d选自下组:阿霉素、表柔比星、柔红霉素、伊达比星、米托蒽醌、长春碱、长春新碱、长春瑞滨、安吖啶、拓扑替康、伊立替康。还优选组合物包括胶体。优选的流体载剂是水或水性介质,在其中,所述药理学活性剂d的硫酸盐/酯或磺酸盐/酯是不可溶或者基本不可溶的。“基本不可溶”应理解为溶解度小于0.1重量%,特别地,小于0.05或0.02重量%。可以将组合物设计成用于动脉内、腹膜内、肌肉内、经皮或静脉给药。可以以任意合适的顺序进行上述步骤。所述药理学活性剂d的所述硫酸盐/酯或磺酸盐/酯的一种优选形式是平均粒度(n)最高至100μm、优选最高至50μm或10μm或5μm的粉末。所述药物组合物的优选形式是水性悬液,特别是水性胶体。根据本发明,还揭示了本发明的药物组合物的生产方法,该方法包括:提供所述抗癌药物与非两亲的无机或有机酸的盐的第一水性溶液;提供第二水性溶液,所述第二水性溶液包含的化学式为(na或k)+(r1so3)-的磺酸烷基酯的钠盐或钾盐或者化学式为(na或k)+(r2oso3)-的硫酸链烷酯的钠盐或钾盐的量等于所述盐的量;混合所述第一和第二溶液。虽然可以在该方法中使用除了钠和钾之外的盐,但是使用它们不是优选的。优选地,r1是直链c6-c30烷基;r2是直链c6-c30烷基;n是1-4的整数。r1和r2优选是直链c10-c20烷基,更优选是直链c12-c18烷基,最优选约为直链c16烷基。根据本发明,还揭示了包含抗癌试剂的本发明的组合物用于靶向递送到人体或动物的肝脏,所述抗癌试剂选自下组:阿霉素、表柔比星、柔红霉素、伊达比星、米托蒽醌、长春碱、长春新碱、长春瑞滨、安吖啶、拓扑替康、伊立替康。根据本发明,还揭示了包含抗癌试剂的本发明的两亲颗粒磺酸盐/酯粉末用于制造包含能够靶向递送到人体或动物的肝脏的药物组合物的药剂,所述抗癌试剂选自下组:阿霉素、表柔比星、柔红霉素、伊达比星、米托蒽醌、长春碱、长春新碱、长春瑞滨、安吖啶、拓扑替康、伊立替康。下面将通过本发明的多个非限制性实施例来更详细阐述本发明。附图简要说明图1显示阿霉素硫酸烷基酯在30%水性乙醇中的溶解度对于烷基链长的依赖性;图2显示阿霉素磺酸链烷酯在30%水性乙醇中的溶解度对于烷基链长的依赖性;图3显示米托蒽醌硫酸烷基酯在30%水性乙醇中的溶解度对于烷基链长的依赖性;图4显示米托蒽醌磺酸链烷酯在30%水性乙醇中的溶解度对于烷基链长的依赖性;图5显示伊立替康磺酸链烷酯在10%水性乙醇中的溶解度对于烷基链长的依赖性;图6显示长春瑞滨磺酸链烷酯在20%水性乙醇中的溶解度对于烷基链长的依赖性;图7显示wistar大鼠注入一剂2小时之后的肝脏中阿霉素浓度对于阿霉素链烷硫酸酯和磺酸酯在30%水性乙醇中的溶解度的依赖性;图8显示阿霉素的非共价键合络合物在30%水性乙醇中的溶解度与加州兔肝脏静脉注射0.5小时之后的阿霉素浓度增加之间的关系;图9显示在静脉注射5mg/kg水性阿霉素盐酸盐和1.25mg/kg的根据实施例1的本发明的组合物之后的肝脏和心脏中的阿霉素浓度。(图1-6的)指数趋势线以及(图7和8)的对数趋势线以及它们的等式是通过微软excel软件的方式获得的。具体实施方式一般过程:材料和方法通过如下方式确定在水性乙醇中的溶解度:将充分量的新鲜获得的胶体以3000rpm离心30-90分钟,滗析上清液,添加10ml的水并振荡混合物,然后重复离心,振荡和清洗3次。来自最终离心的离心液在室温下空气干燥72小时,之后真空干燥24小时。一部分的经过干燥的离心液(20mg)通过搅拌再悬浮于6ml的水性乙醇中,处于室温下持续24小时。混合物在3000rpm离心10分钟,以及上清液过滤通过0.2微米过滤器以去除未溶解固体产物的团聚物。通过uv法确定化合物的溶解度。用于体内研究的组合物是新鲜制备或者通过浓缩物的稀释获得。对于体内研究,选择年龄为60-75天、体重为300g±30g的雌性wistar大鼠。使用4个动物用于测试一种组合物。以5mg/kg阿霉素的量,通过单剂注入尾部给药阿霉素组合物。在死亡之后,立即将大鼠体深冻入液氮中。确定阿霉素在肝脏组织中的生物分布。从肝脏的不同部分,选取5或6片总重约为1g的肝脏组织。样品用含有hcl的水性乙醇溶液以7000rpm均质化20s,以及以11000rpm均质化10s。均化物涡旋30分钟,以及以3000rpm离心30分钟。用一氯乙酸溶液处理上清液并孵育1小时。混合物以15000rpm离心15分钟。通过荧光分析确定最终上清液中的阿霉素浓度。实施例1:制备胶体阿霉素硫酸烷基酯和磺酸链烷酯在室温下,向锥形烧瓶中的5%水性右旋糖中的阿霉素盐酸盐溶液(50ml,1mg/ml)添加与阿霉素盐酸盐相同的溶剂中的5-10摩尔%过量的na+(r1so3)-或na+(r2oso3)-的溶液。作为5%水性右旋糖的替代,可以在该实施例和其他实施例中使用林格氏溶液或者0.9%的盐水或磷酸盐缓冲盐水或者270-300mosm/l的克分子渗透压重量浓度的其他水性溶液。目测监测胶体形成过程。在完成添加之后,混合物涡旋或振荡从30分钟到7天的额外时间段。然后胶体直接使用或者存储在冰箱中。通过uv法,以495或233nm确定组合物中阿霉素的浓度。对于取样,用甲醇稀释胶体的等分试样(甲醇过量>20:1)。实施例2:制备胶体米托蒽醌硫酸烷基酯和磺酸链烷酯在室温下,向锥形烧瓶中的5%右旋糖水溶液中的米托蒽醌二盐酸盐的剧烈搅拌溶液(40ml,0.2mg/ml)添加如下溶液,所述溶液含有与米托蒽醌二盐酸盐相同的溶剂中的0.03毫摩尔%的na+(r1so3)-或na+(r2oso3)-。目测监测黑色胶体的形成。胶体缓慢分解成黑色沉淀物和灰白色上清液。在完成添加之后,获得的混合物搅拌一段额外时间(1-7天)。胶体组合物直接使用或者存储在冰箱中以待后用。通过uv法,以662、611或242nm确定胶体中米托蒽醌的浓度。对于取样,用甲醇稀释胶体的等分试样至>20:1。实施例3:制备胶体伊立替康硫酸烷基酯和磺酸链烷酯在室温下,向去离子水中的伊立替康盐酸盐三水合物的剧烈溶液(5ml,4mg/ml)中添加含有去离子水中的na+(r1so3)-或na+(r2oso3)-的溶液。目测监测胶体的形成。在完成添加之后,获得的混合物搅拌2天,以及混合物以3000rpm离心10分钟。静置时,白色胶体缓慢分解成白色沉淀物和近乎无色的上清液。用5%的水性右旋糖替代上清液。通过涡旋10分钟,使得沉淀物重悬浮在水中。然后,获得的组合物直接使用或者存储在冰箱中以待后用。通过uv法,以360、255或220nm确定胶体或改性胶体中伊立替康的浓度。对于取样,用甲醇稀释产物的等分试样(甲醇过量>20:1)。实施例4:制备胶体长春瑞滨硫酸烷基酯和磺酸链烷酯在室温下,向锥形烧瓶中的5%水性右旋糖中的长春瑞滨酒石酸盐的剧烈搅拌溶液(2ml,5mg/ml)添加与长春瑞滨酒石酸盐相同的溶剂中的一当量的na+(r1so3)-或na+(r2oso3)-的溶液。目测监测胶体的形成。在完成添加之后,获得的混合物涡旋或振荡7天。静置时,胶体缓慢分解成沉淀物和透明上清液。胶体直接使用或者存储在冰箱中以待后用。通过uv法,以268或212nm确定胶体中长春瑞滨的浓度。对于取样,用甲醇稀释胶体的等分试样(甲醇过量>20:1)。实施例5:胶体阿霉素硫酸烷基酯和磺酸链烷酯在30%水性乙醇中的溶解度根据材料和方法中所述的一般方法确定溶解度。结果总结见表1并如图1和2所示。表1:胶体阿霉素硫酸烷基酯和磺酸链烷酯在30%乙醇中的溶解度(v/v)实施例6:胶体米托蒽醌硫酸烷基酯和磺酸链烷酯在30%水性乙醇中的溶解度根据材料和方法中所述的一般方法确定溶解度。结果总结见表4并如图3和4所示。表2:胶体米托蒽醌硫酸烷基酯和磺酸链烷酯在30%乙醇中的溶解度(v/v)实施例7:伊立替康磺酸链烷酯在10%水性乙醇中的溶解度根据材料和方法中所述的一般方法确定溶解度。结果总结见表3和图5。表3:胶体伊立替康磺酸链烷酯在30%乙醇中的溶解度(v/v)烷基链长度,n溶解度,mg/ml100.83483120.15453140.07064160.01476实施例8:胶体长春瑞滨磺酸链烷酯在20%水性乙醇中的溶解度根据材料和方法中所述的一般方法确定溶解度。结果总结见表4和图6。表4:胶体长春瑞滨磺酸链烷酯在20%水性乙醇中的溶解度烷基链长度,n溶解度,mg/ml101.11868120.25692140.06731160.01977实施例9:阿霉素在wistar大鼠肝脏的分布的体内分析将阿霉素盐酸盐的水性溶液、阿霉素硫酸链烷酯的水性胶体、以及阿霉素磺酸烷基酯的水性胶体经由单剂注射给药到尾部中。动物在经由单剂注射给药到尾部中之后2小时死亡;总阿霉素剂量为5mg/kg。根据上文所述过程确定肝脏中的阿霉素浓度。结果总结见表5和图7。表5:阿霉素在wistar大鼠肝脏中的分布的体内分析实施例10:阿霉素在加州兔肝脏中的分布的体内分析以1ml/分钟的缓慢流动速率将阿霉素和阿霉素盐酸盐的胶体非共价络合物给药到加洲兔的边缘耳静脉,阿霉素的总剂量是5mg/kg。动物在给药后0.5小时死亡。根据上文所述过程确定肝脏中的阿霉素浓度。结果参见表6和图8。表6:加洲兔肝脏中的阿霉素分布实施例11:制备包含在30%水性乙醇中具有所需溶解度的阿霉素磺酸盐/酯的胶体该实施例阐述了在特定溶剂中制备具有所需溶解度的本发明的胶体。设定如下参数:活性剂:阿霉素;阴离子:具有偶数数量的碳原子的磺酸链烷酯;溶剂:30%水性乙醇;所需溶解度:0.1mg/ml。基于中等链长的磺酸链烷酯基团中的碳原子数量是相加的假设进行计算。还基于30%水性乙醇中的溶解度y的连续函数的假设来进行计算。根据由实施例5(参见图2)获得的函数(f1),表示了碳原子数量x与溶解度y之间的关系。y=f1(x)=1754.71710exp(-0.74429x)(方程式1)函数(f2)进行逆计算,即从给定溶解度y确定碳原子数量x:x=f2(y)=-1/0.7442855697ln(y/1754.71709855)(方程式2)对于y=0.1mg/ml的溶解度,函数f2得出x等于13.20628。基于基团中碳原子数量的相加效应的假定,c12和c14磺酸盐/酯的比例行为(磺酸盐/酯对具有最接近所需情况的溶解度),提供了所示的c13.20628基团:一当量的c13.20628等于0.397当量的c12和0.603当量的c14的混合物。根据实施例1所述的一般方法来制备具有确定的c12和c14比例的阿霉素磺酸盐/酯的混合物。根据上文所述的一般方法确定溶解度,溶解度是0.098713mg/ml。实施例12:本发明的组合物静脉靶向递送到加洲兔的肝脏通过注入耳静脉,将根据实施例1的本发明的阿霉素组合物(r=c16磺酸盐/酯)给药到加洲兔,浓度为1.25mg阿霉素/kg体重。作为对比,以相同方式给药水性阿霉素盐酸盐,浓度为5.0mg阿霉素/kg体重。如图9所示,对于将阿霉素递送到肝脏,本发明的组合物比现有技术阿霉素盐酸盐更为有效4倍,而对于递送到心脏而言没有差别。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 含叔胺药物物质的靶向递送的制造方法与工艺
- 一种磁靶向双载药递释系统及其制备方法与流程
- 用于引导和靶向按需物质递送的纳米结构的载体的制造方法与工艺
- 附载蟾毒灵的多肽修饰的聚甲基丙烯酸寡聚乙二醇酯-聚己内酯纳米制剂的制作方法
- 附载蟾毒灵的多肽修饰的聚甲基丙烯酸寡聚乙二醇酯-聚己内酯纳米制剂的制作方法
- 一种用于治疗去势抵抗性前列腺癌的靶向前药及其纳米制剂和制备方法
- 一种具有内涵体逃逸功能的肿瘤靶向前药及其纳米制剂和制备方法
- 具有脑肿瘤靶向和肿瘤组织穿透能力的d构型多肽及其基因递释系统的制作方法
- 聚乙二醇-聚乳酸羟基乙酸-聚赖氨酸纳米递送系统、制备方法及其应用的制作方法
- 载蟾蜍灵的环肽修饰的聚乙二醇-聚乳酸羟基乙醇酸-聚赖氨酸纳米粒的制作方法
- 靶向PD-1的多肽及其应用的制造方法与工艺
- 靶向同位素生产系统的制造方法与工艺
- 在制备68GA‑螯合物官能化的靶向剂中用作金属抑制剂的单糖、二糖或多糖的制造方法与工艺
- 用于引导和靶向按需物质递送的纳米结构的载体的制造方法与工艺
- 通过PROSTRATIN靶向K‑RAS介导的信号转导通路和恶性肿瘤的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺