ANGPT2分泌抑制剂在制备用于治疗血管瘤的药物中的应用的制作方法
本发明属于生物医药技术领域,尤其涉及ANGPT2分泌抑制剂在制备用于治疗血管瘤的药物中的应用。
背景技术:
脑海绵状血管瘤(cerebral carvernous malformation,CCM)是发生在中枢神经系统的一种血管错构畸形,是先天性脑血管畸形的一种,约占脑血管畸形的10%~20%,人群患病率约0.1%~4%。该病临床症状隐袭,主要表现为头痛、痫性发作、颅内出血和局部神经功能障碍。但是由于CCM发病机制尚不清楚,目前缺乏有效的治疗药物,手术切除是CCM的唯一治疗手段,因此,寻找CCM发病的关键基因,阐明其调控机制,对揭示CCM疾病发生的本质、确立药物靶点,从而进一步达到有效防治CCM及其他血管发生异常的疾病的目的,具有重要的意义。
血管生成素(angiopoietin)是内皮细胞特异性的促血管生成因子,在血管生成中起到重要作用。血管生成素家族有4种亚型,分别为Ang-1,Ang-2,Ang-3,Ang-4。Ang-1为由498个氨基酸组成的同源六聚体,基因位于8q22,主要由血管内皮周围细胞(如周细胞)分泌,广泛分布于胚胎及富含血管的成熟组织中,如子宫内膜、卵巢、肺、皮肤等。Ang-2基因位于8q23,是由496个氨基酸组成的同源二聚体,主要由内皮细胞分泌,分布于胚胎及血管重塑明显的成熟器官,如胎盘、子宫、卵巢等。Tie 2是血管生成素家族的共同受体,Ang-1可与Tie 2受体特异性结合使其磷酸化,调节血管内皮细胞间及和周围支持细胞间的相互作用,促进微血管结构的成熟和稳定,降低血管通透性。Ang-2(又简称ANGPT2)通过竞争阻断Ang-1与Tie 2受体结合,抑制Tie 2受体磷酸化,减弱内皮细胞与周围支持细胞及基质间的相互作用,解除血管基底膜和周围细胞对血管结构的限制,松解血管结构,促使内皮细胞分离和迁移,导致血管的稳定性降低,通透性增加。因此ANGPT2是CCM的关键基因,可以作为CCM疾病治疗的药物靶点。
胞吐作用是指细胞将含内容物的分泌小泡定向运输释放到细胞外基质的过程。血管内皮细胞包含Weibel-Palade小体(WPB),一种在炎症期间能释放血管性血友病因子(vWF)和P-selectin的分泌型囊泡。这些膜结合囊泡含有可被分泌到细胞外环境中的可溶性蛋白质,在ECs中血管生成素-2(ANGPT2)就是从WPBs释放,进一步结合酪氨酸激酶受体TIE-2,在血管生成过程中协同调节内皮细胞粘合连接蛋白依赖性的血管稳定和血管形成。胞吐作用是通过可溶性NSF的附着蛋白受体(SNARE)复合物的装配介导分泌囊泡与质膜的融合完成的。膜融合之前,其他蛋白介导和调控囊泡和受体膜之间的初始互动。它们包括质膜蛋白syntaxins、突触相关蛋白(SNAPs)、囊泡相关膜蛋白(VAMPs)、小GTP酶的Rab家族蛋白、exocyst、以及众多的其它调控蛋白。而胞吐是ANGPT2发挥作用的必要步骤,因此,抑制ANGPT2从脑血管内皮细胞的Weibel-Palade小体释放将成为CCM疾病药物开发的一个新方向。
技术实现要素:
一方面,本发明的目的在于克服现有技术存在的不足之处而提供了ANGPT2分泌抑制剂在制备用于治疗血管瘤的药物中的应用,本发明为CCM等血管瘤新药的设计提供了新的靶点,为血管瘤的治疗提供了新方法和新思路。
本发明采用的技术方案为:ANGPT2分泌抑制剂在制备用于治疗血管瘤的药物中的应用,所述血管瘤以单层扩张的血管内皮细胞和减少的周细胞覆盖为特征。
作为对上述技术方案的进一步改进,所述血管瘤为脑海绵状血管瘤。
作为对上述技术方案的更进一步改进,所述脑海绵状血管瘤由CCM3基因的功能缺失引发。
作为对上述技术方案的进一步改进,所述ANGPT2分泌抑制剂通过调控胞吐调控蛋白而抑制血管内皮细胞的胞吐作用来用于治疗血管瘤。
作为对上述技术方案的进一步改进,所述ANGPT2分泌抑制剂包括通过启动子水平抑制胞吐调控蛋白的转录的抑制剂、RNA干扰方法抑制胞吐调控蛋白基因表达的siRNA序列、基于胞吐调控蛋白抗原的抗体、胞吐调控蛋白基因的抑制基因激活剂、抑制胞吐调控蛋白基因表达的蛋白的激活剂、胞吐调控蛋白基因转录后基因表达调控的microRNA的激活剂、促进胞吐调控蛋白基因表达的因子及蛋白的抑制剂、胞吐调控蛋白下游基因的抑制剂。
作为对上述技术方案的更进一步改进,所述胞吐调控蛋白包括质膜蛋白syntaxins、突触相关蛋白SNAPs、囊泡相关膜蛋白VAMPs、小GTP酶的Rab家族蛋白、exocyst、UNC家族蛋白中的至少一种。
作为对上述技术方案的更进一步改进,所述胞吐调控蛋白为UNC13B、VAMPs中的至少一种。
作为对上述技术方案的更进一步改进,所述VAMPs为VAMP3。
作为对上述技术方案的进一步改进,所述ANGPT2分泌抑制剂为抑制UNC13B基因或VAMP3基因表达的siRNA。
具体,所述UNC13B基因的DNA序列如SEQ ID NO.1所示(Gene Bank:NM_006377.3)。
所述UNC13B基因编码的蛋白质序列如SEQ ID NO.2所示(Gene Bank:NP_006368.3)。
具体地,所述VAMP3基因的编码序列详见Gene Bank:AAH07050.1
所述VAMP3基因编码的蛋白质序列详见Gene Bank:AAH07050.1。
另一方面,本发明还提供了与ANGPT2分泌相关的胞吐调控蛋白基因及其表达产物在筛选或制备用于治疗血管瘤的药物中的应用,所述血管瘤以单层扩张的血管内皮细胞和减少的周细胞覆盖为特征。
作为对上述技术方案的进一步改进,所述血管瘤为脑海绵状血管瘤。
作为对上述技术方案的更进一步改进,所述脑海绵状血管瘤由CCM3基因的功能缺失引发。
作为对上述技术方案的进一步改进,所述胞吐调控蛋白基因包括质膜蛋白syntaxins基因、突触相关蛋白SNAPs基因、囊泡相关膜蛋白VAMPs基因、小GTP酶的Rab家族蛋白基因、exocyst基因、UNC家族蛋白基因中的至少一种。
作为对上述技术方案的更进一步改进,所述胞吐调控蛋白基因为UNC13B基因、VAMPs基因中的至少一种。
作为对上述技术方案的更进一步改进,所述VAMPs基因为VAMP3基因。
又一方面,本发明还提供了UNC13B基因、VAMPs基因及其表达产物在筛选或制备用于治疗与ANGPT2分泌相关的疾病中的应用。
作为对上述技术方案的进一步改进,所述与ANGPT2分泌相关的疾病为脑海绵状血管瘤。
作为对上述技术方案的更进一步改进,所述脑海绵状血管瘤由CCM3基因的功能缺失引发。
作为对上述技术方案的进一步改进,所述VAMPs基因为VAMP3基因。
CCM3敲除的的HBMVECs能显著增加胞吐作用,但不影响囊泡与质膜融的动力学;然而,同时沉默UNC13B在HBMVECs表达能抑制因CCM3敲低引起的增多的胞吐作用。在脑ECs敲低CCM3能明显增强细胞ANGPT2的分泌,该作用能被同时沉默UNC13B或VAMP3所阻断。因此,可以通过合成或制作抑制UNC13B基因转录载体和试剂,或抑制UNC13B基因表达的siRNA序列,或者基于UNC13B抗原蛋白的抗体,或用于抑制UNC13B蛋白质活性的其它试剂用作基因治疗,也可根据该基因所抑制的下游基因功能,制作出其下游基因的抑制物(靶向治疗药物),制备出治疗CCM及其他血管畸形疾病药物。
为了直接检测CCM3是否在ECs调控胞吐作用,我们把VAMP8与pH敏感的荧光蛋白PHluorin融合,用活细胞TIRF显微镜观察一个个胞吐囊泡颗粒融合的过程。我们发现CCM3与囊泡相关膜蛋白(VAMP3和VAMP8)共定位。在脑ECs敲低CCM3(而不是CCM1或CCM2)能明显增强细胞ANGPT2的分泌,该作用能被同时沉默UNC13B或VAMP3所阻断。
相对于现有技术,本发明的有益效果为:
发明人的研究结果证明了CCM3缺失或敲除的血管内皮细胞及动物模型中ANGPT2的胞吐作用增强和胞外分泌增加,导致内皮细胞连接的破坏和CCM病变的形成。ANGPT2从脑血管内皮细胞的Weibel-Palade小体释放是CCM疾病药物开发的一个新方向。发明人的研究结果进一步证实ANGPT2从内皮细胞的释放受到UNC13B和VAMP3等胞吐调控蛋白的调控。因此,抑制UNC13B和VAMP3等胞吐调控蛋白功能的药物和手段可能成为CCM及其他血管畸形疾病的治疗药物。
附图说明
图1显示了诱导型的内皮细胞特异性CCM3敲除的小鼠快速出现脑和视网膜CCM病变表型;野生型(WT)和诱导型的内皮细胞特异性CCM3敲除(Ccm3-iecKO)的小鼠在出生后1-3天喂他莫昔芬,在P2-P10天取脑组织进行检测;其中,A显示了P2,P5,P10天的WT和Ccm3-iecKO小鼠脑组织切片HE染色结果;B显示了P2,P5,P10天的WT和Ccm3-iecKO小鼠脑组织总CCM病灶数量统计结果;C显示了P2,P5,P10天Ccm3-iecKO小鼠脑组织CCM不同大小病灶数量统计结果;图D-G显示了CCM病变部位血管渗漏,其中:D为P10天的WT和Ccm3-iecKO小鼠新鲜脑组织;E-G:P10天的小鼠用FITC-葡聚糖灌注后进行免疫荧光检测,E为脑组织荧光图,箭头指示血管渗漏,F为CD31染色图,箭头表示正常脑组织结构,星号*显示CCM病灶,G伊文思蓝白蛋白检测评价小鼠血脑屏障渗透性;图H-J显示了Ccm3-iecKO小鼠视网膜病变,对P10天的WT和Ccm3-iecKO小鼠视网膜组织的CD31和isolectin蛋白进行免疫荧光染色,放大倍数H为20X,I为80X,星号*显示CCM病变部位呈海绵状异常血管团,J显示了损害区域占比。
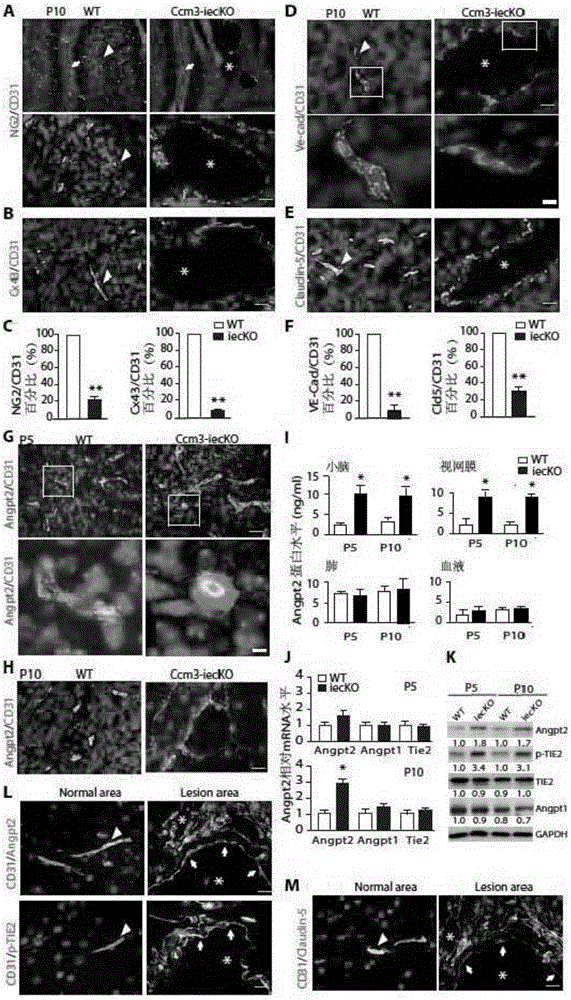
图2显示了Ccm3-iecKO小鼠血管内皮细胞连接的破坏、EC与PC的解离、和ANGPT2表达的升高;野生型(WT)和诱导型的内皮细胞特异性CCM3敲除(Ccm3-iecKO)的小鼠在出生后1-3天喂他莫昔芬,在P5和P10天取脑组织进行检测;其中,A显示了P10天的WT和Ccm3-iecKO小鼠小脑组织切片内皮细胞标记CD31和周细胞标记NG2免疫荧光染色,B显示了CD31和EC–PC缝隙链接标记connexin-43(CX43)染色,其中,箭头指示正常脑组织结构,星号*显示CCM病灶,箭指示小脑叶之间的血管;C分别显示了NG2和CX43在CD31阳性的血管上的覆盖率;D–F显示了P10天的WT和Ccm3-iecKO小鼠小脑组织切片VE-cadherin/CD31染色(D)和claudin-5/CD31染色(E),箭头指示正常脑组织结构,星号*显示CCM病灶;方框区域图片用高倍显示WT和Ccm3-iecKO病灶中VE-cadherin/CD31免疫荧光染色;F分别显示了VE-cadherin和claudin-5在CD31阳性的血管上的覆盖率;G-H显示了P5和P10天的WT和Ccm3-iecKO小鼠小脑组织切片ANGPT2/CD31染色结果,其中图G中的高倍图源自方框区域,标尺:100μm;I显示了ELISA检测WT和Ccm3-iecKO小鼠的小脑、视网膜、肺和血液中的ANGPT2蛋白水平的结果;J-K显示了qRT-PCR和蛋白免疫印迹分别检测P5和P10天的WT和Ccm3-iecKO小鼠小脑组织中ANGPT2-TIE2信号通路的mRNA水平和蛋白水平的结果,所得统计数据为倍数变化(Ccm3-iecKO组数值除以WT组数值所得倍数改变,把WT组的数值当为1);L-M显示了ANGPT2-TIE2信号通路在人CCM3病灶高表达,人CCM3切片标本作CD31和ANGPT2或p-TIE2(L)或Claudin-5(M)免疫荧光共染色,其中,箭头指示正常血管,星号*指示CCM病灶,箭显示CCM病灶中ANGPT2-TIE2染色阳性但Claudin-5染色阴性。n=10,*P<0.05,**P<0.01(非配对双尾t检验),数值为均数±标准误。
图3显示了CCM3抑制血管内皮细胞中ANGPT2分泌和维护血管内皮细胞连接;其中,A显示了免疫共沉淀检测结果,发现UNC13B和CCM3及GCKIII家族成员STK24形成一个复合体;B显示了活细胞TIRF显微镜观察结果,发现CCM3与囊泡相关膜蛋白(VAMP3和VAMP8)共定位;C显示了实时动态自动拍摄的TIRF显微镜记录一个个囊泡与质膜融合的过程及其数量及动力学统计结果,n=10,**P<0.01;D-G显示了CCM3调控血管内皮细胞中ANGPT2分泌;siRNAs转染HBMVECs72小时后,用或不用放线菌酮(CHX)处理2-8小时,ELISA检测培养液中ANGPT2蛋白水平的结果如图D,E,G所示;F显示了蛋白免疫印迹检测分泌出去的和细胞内ANGPT2蛋白水平的结果;H-L显示了CCM3-ANGPT2调控EC细胞连接和EC渗透性,其中,图H–K显示了免疫荧光染色及定量分析细胞粘附连接蛋白VE-cadherin和紧密连接蛋白ZO-1的结果,图L显示了TEER检测细胞屏障功能的结果;需要说明的是,图C、E分别从左至右依次显示了siCtrl、siUNC13B、siCCM3、siCCM3/siUNC13B的融合事件/细胞、融合持续时间和Angpt2释放,图D从左至右依次显示了siCtrl、siCCM1、siCCM2、siCCM3的Angpt2释放,图I、K、L分别从左至右依次显示了siCtrl、siUNC13B、siCCM3/siUNC13B、siCCM3、siCCM3/IgG、siCCM3/αAngpt2的破坏的粘附连接,破坏的紧密连接和跨内皮通量。
图4显示了CCM3维护EC管腔形成和周细胞募集;其中,A-B显示了Organotypic血管生成实验结果;siRNAs转染的HBMVECs被种在单层融合的成纤维细胞之上,在培养液中加或不加anti-ANGPT2(10μg/ml),共培养14天,通过VE-cadherin和collagen IV免疫荧光染色观察EC出芽和管腔形成过程,结果如图A所示;B显示定量分析显示血管分支的数量、平均管腔直径、和成管的面积(collagen IV覆盖的区域);C-D显示了3D出芽实验结果;siRNAs转染的HBMVECs包被在cytodex球珠上,然后cytodex球珠被埋在fibrin胶里,在EGM2培养液中培养8天,结果如图C、D所示,其中,箭头指示出芽,星号指示管腔,图D显示了出芽数量、出芽长度和管腔面积的定量分析结果;图E-F显示了3D出芽实验观察EC-PC相互作用;为了同时观察出芽过程中的EC和PC,我们把表达绿色荧光蛋白EGFP的HBMVEC转染不同的siRNAs(包括Ctrl或CCM3或UNC13B)和表达红色荧光蛋白mCherry的正常HBMVPC以2:1的比例同时包被在cytodex球珠上,在培养液中加或不加anti-ANGPT2(10μg/ml),共培养8天;图E、F显示了观察到的EC出芽和周细胞覆盖率及定量分析结果;n=10,*P<0.05,**P<0.01,标尺:100μm,实验重复三次;需要说明的是,图B从左至右依次显示了siCtrl、siUNC13B、siCCM3、siCCM3/siUNC13B、siCCM3/αAngpt2的分支数量、管腔直径、血管面积,图D从左至右依次显示了siCtrl、siUNC13B、siCCM3、siCCM3/siUNC13B、siCCM3/αAngpt2的出芽数量、出芽长度、血管面积,图F从左至右依次显示了siCtrl、siUNC13B、siCCM3、siCCM3/siUNC13B、siCCM3/αAngpt2、siCCM3/siUNC13B/αAngpt2的出芽数量、PC覆盖率。
图5显示了Unc13b基因敲除能减少Ccm3-iecKO小鼠的CCM病变的形成;野生型(WT)、Unc13b–/–、诱导型的内皮细胞特异性CCM3敲除(Ccm3-iecKO)和在Ccm3-iecKO基础上合并Unc13b基因敲除(Ccm3lox/lox:Cdh5-CreERT2:Unc13b-/-,DKO)的小鼠在出生后1-3天喂他莫昔芬,在P10天取脑组织进行检测;图A显示了新鲜脑组织大体形态学图片,箭显示CCM病灶,标尺:2mm;图B-C显示了HE染色和CC M病灶统计结果;图D-E显示了NG2/CD31免疫荧光共染和定量分析结果;图F-G显示了ANGPT2/CD31和p-TIE2/CD31免疫荧光共染及定量分析结果;图H-I显示了ELISA和蛋白免疫印迹检测脑组织中ANGPT2蛋白水平的结果;图J显示了分离小鼠的小脑的血管内皮细胞并用ELISA检测脑组织中ANGPT2蛋白水平的结果;n=10,*P<0.05,**P<0.01,标尺:A:2mm;B(上图):400μm;B(下图),D,F:100μm,数值为均数±标准误;需要说明的是,图C、E、G、H、J分别从左至右依次显示了WT、Unc13b-/-、Ccm3-iecKO、DKO的CC M病灶数量、NG2覆盖率、p-TIE2阳性的EC、Angpt2蛋白水平、Angpt2分泌量。
具体实施方式
为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。实施例中未进行详细描述的方法均可通过常规试验操作方法实施。
它莫西芬(tamoxifen),也即他莫昔芬;Ccm3与CCM3可替换使用;Ang-2、Angpt2、ANGPT2可替换使用;磷酸化的TIE2、p-Tie2、p-TIE2可替换使用;Unc13b和Unc13B可替换使用;Px天即第x天;αAngpt2即Angpt2中和抗体。
实施例
1、材料与方法
1.1 它莫西芬诱导的血管内皮细胞特异性的Ccm3基因敲除鼠(Ccm3-iecKO)模型的建立
为了便于通过Cre-LoxP系统研究Ccm3在血管内皮细胞中的功能,我们使用Cdh5-CreERT2转基因小鼠。Cdh5-CreERT2转基因技术原理是,首先将雌激素受体(ER)的配体结合域(LBD)突变,进而使其与Cre重组酶融合产生一个嵌合重组酶(Cre-ER),该酶活性依赖于它莫西芬的存在,而不受体内雌激素的影响。通过利用Cdh5基因的启动子驱动Cre重组酶基因在血管内皮细胞中的表达。该转基因小鼠若服用它莫西芬,Cre重组酶的活性被激活,即可导致Cre介导的重组在预定时间内和特定的组织细胞类型(这里是指血管内皮细胞)上发生。
Cdh5-CreERT2转基因小鼠的获取也可参考已报道的文献(可参见:Wang,Y.,et al.Ephrin-B2controls VEGF-induced angiogenesis and lymphangiogenesis.Nature.2010May 27;465(7297):483-6.doi:10.1038/nature09002.)。Ccm3fl/fl小鼠(可参见:He Y,et al.Stabilization of VEGFR2signaling by cerebral cavernous malformation 3is critical for vascular development.Sci Signal.2010Apr 6;3(116):ra26.doi:10.1126/scisignal.2000722.)与Cdh5-CreERT2转基因小鼠交配,即得到可诱导的血管内皮细胞特异性的Ccm3基因敲除鼠(Ccm3-iecKO)。为了在小鼠体内用它莫西芬诱导Ccm3基因的敲除,我们用玉米油把它莫西芬(Sigma,T5648)配成浓度为10mg/ml的液体,给刚出生的Ccm3-iecKO和WT(Ccm3fl/fl)幼鼠从P1至P3每天喂10μg/g(体重)的它莫西芬一次,连续三天。我们从Dr.Nils Brose(Max Planck Institute of Experimental Medicine,Germany)得到Unc13B基因缺失鼠,该鼠无任何表型,除了在1年后可发生癫痫,这可能是由于UNC13B在脑的功能缺失导致。
1.2 临床样品
我们从耶鲁大学医学院的神经病理学系的人体标本档案馆得到CCM3样品。所有样品均由两名独立的富有经验的病理学家来评估CCM3样品的免疫组化结果,他们事先并不知道患者的临床信息。我们得到8例CCM3患者的蜡块标本,并且每个蜡块已有切好的切片20多张。我们通过H&E染色发现6例CCM3样本含有典型的CCM样损害,其病变周围有相对正常的组织包绕;2例CCM3样本有明显的血管纤维化。我们进一步把这8例CCM3样本进行免疫染色分析。
1.3 小鼠体内ANGPT2中和抗体治疗
我们用无菌的生理盐水把人源化的鼠单克隆ANGPT2中和抗体(Genentech,USA,或DarronMedscience公司(广州道瑞医药科技有限公司))稀释成1mg/mL。从P2开始,隔天腹腔注射ANGPT2中和抗体(10μg/g体重)或正常IgG1到WT或Ccm3-iecKO小鼠体内。
1.4 异硫氰酸荧光素葡聚糖(FITC-dextran)灌注和伊文思蓝(Evan Blue)染料渗透试验
异硫氰酸荧光素葡聚糖(FD2000S;Sigma-Aldrich公司,St.Louis,MO,美国)溶于无菌的生理盐水,离心10,000g 5分钟,调整浓度为50mg/ml,收集上清,避光。同胎出生的WT或Ccm3-iecKO小鼠经ANGPT2中和抗体处理或不经ANGPT2中和抗体处理均称重,并腹部注射麻醉((氯胺酮100mg/kg+甲苯噻嗪10mg/kg))。眼球后注射按照已发表的文献进行。注射部位选定为左眼角外眦。用3/10ml胰岛素注射器(31G针头)轻轻的沿45°角刺入小鼠的眼眶静脉窦。以10μl异硫氰酸荧光素葡聚糖对应1g体重来根据体重换算相应异硫氰酸荧光素葡聚糖注射量,并注射入小鼠眼眶静脉窦。5分钟后,收集脑和视网膜,并进行组织铺片或切片免疫染色处理。
伊文思蓝染料渗透性实验(Miles实验)。伊文思蓝染液(用0.9%NaCl溶液配成1%伊文思蓝溶液;Sigma-Aldrich)注射入已麻醉的WT和Ccm3-iecKO小鼠眼球后血管丛,注射量为10μl/g体重。注射30分钟后,杀死小鼠并往左心室灌注PBS,以清洗血管内的染料。收集小脑组织,并在60℃中干燥过夜,在伊文思蓝染料取出前称重,用1ml的甲酰胺55℃下孵育小脑组织16小时以取出伊文思蓝染料。用分光光度计(激发光波长为630nm)来检测伊文思蓝的含量。
1.5 免疫荧光分析
收集小鼠脑组织,用4%多聚甲醛固定,OCT包埋并切片(5μm厚度),PBS冲洗两次组织切片以移除OCT,并用PBS配成含5%驴血清和0.3%TritonX-100的封闭液,封闭和渗透组织切片半小时,以排除非特异性染色。组织切片用一抗4℃孵育过夜,如抗CD31(1:100),抗NG2(1:100),抗VE-cadherin(1:100),抗ZO-1(1:100),抗Claudin-5(1:100),抗Angiopoietin-2(1:100),抗Connexin-43(1:100)等。次日用PBS冲洗组织切片三遍,用二抗(1:200to 1:400)在室温下孵育组织切片1小时。然后用PBS冲洗三遍,组织切片用含DAPI(Vector Laboratory)的防荧光淬灭封片剂封片并拍照。细胞爬片免疫荧光染色方法与组织切片类似。简言之,4%PFA固定HBMVECs 20分钟,用PBS清洗掉PFA,继而用0.1%的TritonX-100处理细胞爬片2分钟以增加细胞通透性。封闭细胞半小时,相继用一抗二抗孵育细胞,然后进行封片和拍照。
视网膜荧光染色。异硫氰酸荧光素葡聚糖灌注P5至P15出生的新生小鼠的眼眶周静脉丛,收集眼球并在冰上用4%多聚甲醛固定2小时。继而用PBS清洗掉4%PFA,在显微镜下去除角膜、晶状体、巩膜和玻璃体血管,解剖出完整的视网膜。将视网膜浸润于含5%驴血清、0.5%Triton的PBS中4℃封闭过夜,然后相继用含1%驴血清、0.1%Triton的PBS稀释一抗(1:50)并4℃孵育过夜,荧光二抗4℃孵育过夜,用PBS清洗视网膜5次。以视乳头为中心,在视网膜上用剪刀对称地沿周边视网膜向中心放射状做4个切口,使视网膜平铺成花瓣状,荧光封片剂封片。用TCS SP5共聚焦显微镜(Leica,德国)拍照。用NIH Image J软件统计血管面积占整个视网膜的百分比。
1.6 组织中的基因表达
用含DNase I的RNeasy kit(Qiagen,Valencia,CA)消化组织,提取出总RNA。按Qiagen标准程序(Super Script first-strand synthesis system;Qiagen)将RNA(1μg量)进行反转录。继而用特定引物(如ANGPT1,ANGPT2和Tie-2)和iQ SYBR Green Supermix配好反应液,在iCycler实时监测系统(Bio-Rad Laboratories,Inc.,Hercules,CA)进行实时定量聚合酶链式反应(qPCR)。ANGPT1,ANGPT2和Tie-2引物序列如下:
小鼠来源:
ANGPT1RTF:ATTCTTCGCTGCCATTCT(SEQ ID NO.21)
ANGPT1RTR:CAGTTCCCGTCGTGTTCT(SEQ ID NO.22)
ANGPT2RTF:TTCCTCTTGGGTGCTTTA(SEQ ID NO.23)
ANGPT2RTR:CGCTTATTTGGTGGTTATT(SEQ ID NO.24)
Tie2RTF:AGGGCAAGATGGATAGGG(SEQ ID NO.25)
Tie2RTR:TGAGCACAGGGAGGGTTA(SEQ ID NO.26)
人来源:
ANGPT1RTF:AGAACCTTCAAGGCTTGGTTAC(SEQ ID NO.27)
ANGPT1RTR:GGTGGTAGCTCTGTTTAATTGCT(SEQ ID NO.28)
ANGPT2RTF:CTCGAATACGATGACTCGGTG(SEQ ID NO.29)
ANGPT2RTR:TCATTAGCCACTGAGTGTTGTTT(SEQ ID NO.30)
Tie2RTF:TCCGCTGGAAGTTACTCAAGA(SEQ ID NO.31)
Tie2RTR:GAACTCGCCCTTCACAGAAATAA(SEQ ID NO.32)
1.7 细胞培养和生长因子
我们根据以前的实验方法(可参见:Waters JP,Kluger MS,Graham M,Chang WG,Bradley JR and Pober JS.In vitro self-assembly of human pericyte-supported endothelial microvessels in three-dimensional coculture:a simple model for interrogating endothelial-pericyte interactions.J Vasc Res.2013;50:324-31.)分离Ccm3-iecKO小鼠脑微血管内皮细胞(Mouse brain microvascular ECs,MBMVECs)。纯的微血管内皮细胞表达VE-cadherin,血管性假性血友病因子(von Willebrand factor)和CD31阳性及VEGFR2信号,但不表达周细胞的标记(如SMA和NG2)。所有实验我们用3代以内的原代脑微血管内皮细胞。我们用100nM的4羟基它莫西芬(Sigma,H7904)处理MBMVECs以在体外诱导Ccm3基因的敲除,用溶剂(DMSO/乙醇)处理的细胞为正常对照组。我们从Angio-Proteomie公司(Boston,USA)购买到人脑微血管内皮细胞(Human brain microvascular ECs,HBMVECs;cAP0002)和人脑微血管周细胞(HBMVPCs;cAP-0030)。内皮细胞用微血管内皮细胞生长液(Microvascular Endothelial Cell Growth Medium-2MV,Lonza)培养,周细胞用含有FBS和生长因子(Angio-Proteomie)的周细胞培养液培养。人肺成纤维细胞用含有10%FBS的DMEM培养液培养,并在14代以内使用。
1.8 慢病毒载体和包装系统
我们使用Trans-Lentiviral包装系统。CCM3-WT(1-212aa)和CCM3-N(人类患者中含有N端1-95-aa的缩短突变体)的编码序列通过pLEX MCS载体克隆(Clontech)。从Clontech购买能表达mCherry(rLV.EF1.mCherry-9)的慢病毒。转染质粒并包装慢病毒。转染62小时后,收集条件培养基,通过低速离心清除碎片,0.45-μM过滤器(Falcon,Lincoln Park,NJ)过滤。然后用所得条件培养基稀释病毒载体,并加入8μg/ml聚凝胺(Sigma,St.Louis,MO),培养过夜转染HBMVEC。第二天换液,细胞继续培养36小时,并用荧光显微镜观察RFP的表达量。CCM3突变的表达量用蛋白质印迹检测。通过50,000g 90分钟高速离心条件培养液来浓缩病毒载体。离心后的浓缩小球用含有4μg/ml聚凝胺的PBS重悬,PBS体积为0.05%的初始条件培养液的容量,滴定病毒载体浓度并于–80℃中保存。
1.9 酶联免疫吸附测定ANGPT2
为测量人内皮细胞的上清液中ANGPT2的含量,处理细胞后在不同时间点收集细胞上清液。ELISA实验严格按照标准的ELISA方法实施,并用以下抗体检测,R&D的抗ANGPT2抗体、重组人ANGPT2(625-AN-025)、人ANGPT2Mab(MAB098)、人ANGPT2生物素化Mab(Bam0981).为了测量在老鼠细胞和组织的ANGPT2,相应的ELISA试剂盒来自R&D(MANG20)和ABCAM(ab171335)。为进行免疫印迹,速冻老鼠组织,并在聚丙烯酰胺凝胶电泳前混匀于RIPA裂解液。为酶联免疫吸附实验,需碾碎新鲜的老鼠组织并在无血清培养皿中孵化2小时,进而测定上清液中的ANGPT2蛋白水平。
1.10 实时TIRF显微镜
我们根据以前的实验方法(可参见:Zhang Y,Tang W,Zhang H,Niu X,Xu Y,Zhang J,Gao K,Pan W,Boggon TJ,Toomre D,Min W and Wu D.A network of interactions enables CCM3and STK24to coordinate UNC13D-driven vesicle exocytosis in neutrophils.Dev Cell.2013;27:215-26.)进行实时全内反射荧光显微镜检测。具体用IX70倒置显微镜像(Olympus),装备一个氩激光谱线(488nm),一个TIRFM冷凝器,一个60×1.45NA TIRF物镜(Olympus)和一个EMCCD镜头(iXon887;AndorTechnology)。通过iQ软件(AndorTechnology)控制成像系统。关于活细胞成像,HBMVEC细胞种入含1.5号玻璃底的35mm培养皿中(MatTek,Ashland,MA),并用EGM-2-MV培养基培养(Lonza,cc-3202)。首先用RNAiMAX(ThermoFisher)转染siRNAs到HBMVEC细胞内。24小时后,用Cytofect-内皮细胞转染试剂盒(Cell Applications Inc.,TF101K)转染VAMP8-Phluorin质粒到细胞内。再经过24小时,细胞放入37℃的孵化室,以200毫秒的间隔获取转染细胞的实时动态图像,进而用MATLAB系统的ImageJ 1.42软件(来源于美国国立卫生研究院)处理分析图像。
1.11 跨内皮通量和TEER测量
跨内皮通量的检测,内皮细胞接种入纤维蛋白包被的96W20idf金电极的ECIS培养皿中(Applied BioPhysics),并用EGM-2培养基(Lonza,cc-3202)培养并转染siRNAs。通过记录72小时内的细胞基质的电阻抗传感来评估内皮细胞的屏障功能。
1.12 Organotypic血管生成实验
我们采用经改进的Organotypic血管生成实验(可参见:Hetheridge C,Mavria G and Mellor H.Uses of the in vitro endothelial-fibroblast organotypic co-culture assay in angiogenesis research.Biochem Soc Trans.2011;39:1597-600.)。用逆转录病毒(MOI 20)转染HBMVECs使其表达绿色荧光蛋白(EGFP),2天后再转染siRNAs到细胞体内;或直接给未转染EGFP的HBMVECs转染siRNAs,1天后消化HBMVECs,并在预先铺上2×104人成纤维细胞并达到融合的24孔板内每孔接种含或不含EGFP表达的经siRNAs转染的8.5×103的HBMVECs。我们每两天给细胞换EGM2培养液,并在2至14天在荧光显微镜下直接通过绿色荧光蛋白观察小管形成,或通过VE-cadherin和collagen IV免疫荧光染色观察小管形成。
1.13 三维出芽实验
用LipofectamineRNAiMAX(Invitrogen)转染siCtrl,siUNC13B,siCCM3或siCCM3与siUNC13B的联合到HBMVECs作用8小时或过夜,接着给HBMVECs换成新鲜的微血管内皮细胞生长液(Microvascular Endothelial Cell Growth Medium-2MV,Lonza)培养。我们参照文献(可参考:Nakatsu MN and Hughes CC.An optimized three-dimensional in vitro model for the analysis of angiogenesis.Methods Enzymol.2008;443:65-82.)进行三维出芽实验。细胞转染24小时后,即被消化并与一定数量的微载体珠(C3275,Sigma)混匀在含有1.5MLEGM2-MV培养液的流式细胞管里,放进培养箱培养,4小时内每隔20分钟轻拍打管壁,以使最终每个微载体珠包被有接近400个细胞。第二天,把这些包被有HBMVECs的微载体珠包埋在2mg/ml的纤维蛋白胶里,继而把20,000/孔的成纤维细胞铺在纤维蛋白胶上层,并给每孔细胞1.0ML含或不含ANGPT2中和抗体(1μg/ml或10μg/ml;美国Genentech公司或广州道瑞医药科技有限公司)的EGM2-MV培养液。每两天给细胞换液并从2至13天通过显微镜观察细胞出芽状况、管腔形成、血管分支形成及融合过程。
1.14 HBMVECs和HBMVPCs共培养系统的三维出芽实验
我们参照文献进行血管内皮细胞和周细胞的相互作用的三维出芽实验(可参见:Waters,J.P.,et al.In vitro self-assembly of human pericyte-supported endothelial microvessels in three-dimensional coculture:a simple model for interrogating endothelial-pericyte interactions.J Vasc Res 50,324-331(2013);Chang,W.G.,Andrejecsk,J.W.,Kluger,M.S.,Saltzman,W.M.&Pober,J.S.Pericytes modulate endothelial sprouting.Cardiovasc Res 100,492-500(2013);Scheppke,L.,et al.Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basem entm em brane.B lood 119,2149-2158(2012).)。首先用逆转录病毒(MOI 20)转染HBMVECs使其表达绿色荧光蛋白(EGFP),用腺病毒(MOI 20)转染HBMVPCs使其表达mCherry。继而用LipofectamineRNAiMAX(Invitrogen)转染siCtrl,siUNC13B,siCCM3或siCCM3与siUNC13B的联合到HBMVECs。细胞转染24小时后,即被消化并按ECs:PCs(2:1)的比例与一定数量的微载体珠(C3275,Sigma)混匀,培养在含有1.5MLEGM2-MV培养液的流式细胞管里,放进培养箱,4小时内每隔20分钟轻拍打管壁,以使最终每个微载体珠包被有接近400个细胞。我们选择ECs:PCs(2:1)的比例是因为周细胞比血管内皮细胞长得快,且在ECs:PCs(1:1)的比例条件下周细胞限制内皮细胞的出芽。第二天,把这些包被有HBMVECs的微载体珠包埋在2mg/ml的纤维蛋白胶里,继而把20,000/孔的成纤维细胞铺在纤维蛋白胶上层,并给每孔细胞1.0ML含或不含ANGPT2中和抗体(1μg/ml或10μg/ml;美国Genentech公司或广州道瑞医药科技有限公司)的EGM2-MV培养液。每两天给细胞换液并从2至8天通过荧光显微镜观察内皮细胞出芽状况、血管内皮细胞形成小管的周细胞的覆盖率。
以上试验中我们所用的siRNA由美国Santa Cruz公司合成,人的UNC13B基因和VAMP3基因的siRNA各自均为三个有效siRNA的混合物。UNC13B基因的siRNA为以下三种siRNA的混合物:
sc-42022A:
正义链序列:GGAUUGCGCUGAAGACUAUtt(SEQ ID NO.3)
反义链序列:AUAGUCUUCAGCGCAAUCCtt(SEQ ID NO.4)
sc-42022B:
正义链序列:GACAUCAAGUCAAGAGUAAtt(SEQ ID NO.5)
反义链序列:UUACUCUUGACUUGAUGUCtt(SEQ ID NO.6)
sc-42022C:
正义链序列:GGAUCGACCUCUCUACAUAtt(SEQ ID NO.7)
反义链序列:UAUGUAGAGAGGUCGAUCCtt(SEQ ID NO.8)
VAMP3基因的siRNA为以下三种siRNA的混合物:
sc-41338A:
正义链序列:GGAUUACUGUUCUGGUUAUtt(SEQ ID NO.9)
反义链序列:AUAACCAGAACAGUAAUCCtt(SEQ ID NO.10)
sc-41338B:
正义链序列:GAAGAGGCCUUCUUAAGUUtt(SEQ ID NO.11)
反义链序列:AACUUAAGAAGGCCUCUUCtt(SEQ ID NO.12)
sc-41338C:
正义链序列:GUGUAUUCCUGACAGUUAAtt(SEQ ID NO.13)
反义链序列:UUAACUGUCAGGAAUACACtt(SEQ ID NO.14)
CCM3基因的siRNA为以下三种siRNA的混合物:
sc-62084A:
正义链序列:GGAUAUAGCUAGUGCAAUAtt(SEQ ID NO.15)
反义链序列:UAUUGCACUAGCUAUAUCCtt(SEQ ID NO.16)
sc-62084B:
正义链序列:CAACCGACUAAUUCAUCAAtt(SEQ ID NO.17)
反义链序列:UUGAUGAAUUAGUCGGUUGtt(SEQ ID NO.18)
sc-62084C:
正义链序列:GGAUUAUUGCCAUCUUACAtt(SEQ ID NO.19)
反义链序列:UGUAAGAUGGCAAUAAUCCtt(SEQ ID NO.20)。
2.结论
2.1 诱导型的血管内皮细胞特异性Ccm3敲除的小鼠产生CCM损害
给刚出生的WT(Ccm3fl/fl)幼鼠和可诱导的血管内皮细胞特异性的Ccm3基因敲除(Ccm3-iecKO)小鼠从P1至P3每天喂他莫昔芬,在P2-P10取脑组织进行检测。结果发现,Ccm3-iecKO小鼠能在小脑快速产生CCM血管损害,即扩张膨胀伴出血的毛细血管,该病损在P5可见、P10急剧增大增多(如图1A-C所示)。没有Ccm3-iecKO能存活超过P15(n>50)。FITC-dextran小鼠体内循环灌注实验显示,FITC-dextran局限于WT小鼠脑和视网膜的毛细血管床内,但在Ccm3-iecKO小鼠FITC-dextran弥散渗漏到小脑扩张的毛细血管床外的周边组织(如图1D–F所示)。Evans Blue染料渗漏实验也证实了Ccm3-iecKO小鼠小脑增强的血管渗漏(如图1G所示)。CCM样静脉血管畸形也在Ccm3-iecKO小鼠视网膜周边血管丛被检测到(如图1H–J所示)。
2.2 CCM损害表现为血管内皮细胞连接的破坏和ANGPT2表达的升高
Ccm3-iecKO小鼠的小脑血管损害以单层扩张的血管内皮和严重减少的周细胞覆盖为特征(如图2A-C所示)。缝隙连接蛋白connexin-43免疫荧光染色在Ccm3-iecKO小鼠也明显减少,这表明血管内皮细胞和周细胞相互作用的完整性受破坏(如图2A-C所示)。类似地,Ccm3-iecKO小鼠血管内皮细胞粘合连接蛋白VE-cadherin和紧密连接蛋白claudin-5在小脑CCM血管损害处表达不规则、明显减少(如图2D–F所示)。血管内皮细胞分泌的ANGPT2被经典地认为是ANGPT1的拮抗剂,ANGPT2能拮抗ANGPT1稳定血管的功能,增加血管内皮细胞通透性。在WT小鼠小脑ANGPT2可在正常血管内皮细胞内检测到,然而它在Ccm3-iecKO小鼠的小脑扩张的血管内和血管病损周围均显著升高(如图2G-H所示)。ELISA也检测到Ccm3-iecKO小鼠的小脑和视网膜显著升高的ANGPT2蛋白水平,但在Ccm3-iecKO小鼠的肺和血样未检测到升高的ANGPT2蛋白水平(如图2I所示),这与CCM血管损害形成部位一致。ANGPT2(而不是ANGPT1或TIE2)的mRNA表达水平在P5天Ccm3-iecKO小鼠的小脑轻度增加,在P10显著增加(如图2J所示)。我们意外地发现Ccm3-iecKO小鼠的小脑增加的TIE2蛋白磷酸化水平(如图2K所示),提示CCM血管损害发展过程中ANGPT2-TIE2信号通路的激活。为证明ANGPT2的上调具有临床相关性,我们检测CCM3病变患者的ANGPT2的表达水平。我们在相对正常区域的脑组织未发现ANGPT2和p-TIE2阳性染色,但在CCM病变区域发现显著升高的ANGPT2和p-TIE2表达,伴血管内皮细胞紧密连接的破坏(如图2L-M所示)。
2.3 CCM3抑制ANGPT2分泌和维护血管内皮细胞连接
我们发现UNC13B和CCM3及GCKIII家族成员STK24形成一个复合体(如图3A所示)。为了直接检测CCM3是否在ECs调控胞吐作用,我们把VAMP8与pH敏感的荧光蛋白PHluorin融合,用活细胞TIRF显微镜观察一个个胞吐囊泡颗粒融合的过程。我们发现CCM3与囊泡相关膜蛋白(VAMP3和VAMP8)共定位(如图3B所示)。此外,我们用实时动态自动拍摄的TIRF显微镜记录一个个囊泡与质膜融合的过程并统计其数量及动力学。CCM3缺失的HBMVECs能显著增加胞吐作用,但不影响囊泡与质膜融合的动力学;然而,同时沉默UNC13B在HBMVECs表达能抑制因CCM3敲低引起的增多的胞吐作用(如图3C所示)。
我们进一步在ECs检测低表达CCM3对ANGPT2表达和分泌的直接作用。我们发现在HBMVEC沉默CCM3,而不是CCM1或CCM2,能显著增加细胞ANGPT2的分泌。同时沉默UNC13B在HBMVECs表达能显著抑制因CCM3敲低导致的ANGPT2的过度释放(如图3D–E所示)。然而,ANGPT2的总蛋白水平(如图3F所示)并不受低表达CCM3或UNC13B而改变。在CCM3敲低的ECs,增加的ANGPT2胞外分泌与增强的TIE2磷酸化表达有关。然而,TIE2和ANGPT1总蛋白表达水平不受影响(如图3F所示)。与之前的研究结果一致,CCM3能稳定GCKIII激酶。在ECs敲低CCM3能降低STK24的表达。我们用放线菌酮(CHX)阻断蛋白质合成进一步检测ANGPT2的释放作用。蛋白质合成抑制剂CHX不影响对照组或CCM3缺失组的内皮细胞8小时内ANGPT2的总蛋白和分泌型蛋白水平(如图3G所示)。这结果跟我们之前的理论一致,即ANGPT2主要是由蛋白质释放产生的。
我们进一步探讨CCM3介导的ANGPT2分泌是否对EC细胞连接和EC完整性有影响。免疫荧光染色发现,低表达CCM3显著破坏细胞粘附连接蛋白VE-cadherin和紧密连接蛋白ZO-1。然而,在HBMVECs同时沉默UNC13B表达或用ANGPT2中和抗体能明显重塑正常的细胞粘附连接和紧密连接(如图3H–K所示)。我们用TEER实验发现,同时沉默UNC13B表达或用ANGPT2中和抗体也能明显重塑CCM3缺失的HBMVECs正常的细胞屏障功能(如图3L所示)。
2.4 CCM3-ANGPT2信号轴调控EC管腔形成和周细胞募集
如我们在Ccm3-iecKO小脑所见CCM3调控血管管腔直径,我们用体外实验EC出芽模型和管腔形成模型分析CCM3对内皮出芽和管腔形成的影响。表达绿色荧光蛋白EGFP和/或siRNAs转染的HBMVECs被种在单层融合的成纤维细胞之上,在7至14天共培养过程中观察管腔形成过程。VE-cadherin和collagen IV免疫荧光染色显示CCM3缺失的内皮细胞出芽和管腔形成明显增加(如图4A所示)。定量分析显示血管分支的数量、平均管腔直径、和成管的面积(collagen IV覆盖的区域)在CCM3敲低组明显增加(如图4A–B所示)。因CCM3缺失引起的增加的EC出芽和扩大的管腔形成不受正常IgG处理的影响,但能被同时沉默UNC13B表达或用ANGPT2中和抗体所抑制(如图4A–B所示)。
为了进一步证实CCM3调控EC管腔形成和周细胞募集,我们用3D出芽实验来验证,该实验中ECs包被在cytodex球珠上,然后cytodex球珠被埋在fibrin胶里,继而在胶上层铺上成纤维细胞。我们发现,因CCM3敲低引起的增加的EC出芽和扩大的管腔形成能被同时沉默UNC13B表达或用ANGPT2中和抗体所抑制(如图4C–D所示)。
3D出芽实验也是被用来研究周细胞或平滑肌细胞募集和血管成熟的一个系统。为了同时观察出芽过程中的EC和PC,我们把表达绿色荧光蛋白EGFP的HBMVEC转染不同的siRNAs(包括Ctrl或CCM3或UNC13B)和表达红色荧光蛋白mCherry的正常HBMVPC以2:1的比例同时包被在cytodex球珠上。在出芽过程中,PC被招募到正常的EC上,但CCM3敲低的EC不能招募PC(如图3E所示)。然而,在EC同时沉默UNC13B表达或用ANGPT2中和抗体或两者的结合能恢复正常的PC募集到出芽EC上(如图4E–F所示)。我们的结果提示CCM3能通过抑制EC中ANGPT2的胞吐作用,维护正常EC细胞连接、EC管腔形成和PC募集。
2.5 Unc13b基因敲除能减少Ccm3-iecKO小鼠的ANGPT2的分泌和CCM病变的形成
为了检测是否抑制过高水平的胞吐作用能治疗Ccm3-iecKO小鼠的小脑的CCM损害,我们把Unc13b基因敲除小鼠(Unc13b-/-)与Ccm3-iecKO小鼠(Cdh5-CreERT2:Pdcd10fl/fl)配种得到Ccm3-iecKO:Unc13b-/-(DKO)小鼠,继而从P1到P3给予小鼠喂养它莫西芬,从而得到Ccm3和Unc13b两种基因同时敲除的小鼠。我们在P5(开始出现表型的早期)、P10(血管损害表型严重期)分别取脑组织观察大体形态学和作HE染色。我们在Unc13b-/-小鼠未发现血管损害表型,因Ccm3基因敲除诱导产生的CCM血管损害的数量,扩张膨胀的毛细血管伴随周细胞覆盖的减少(如图5A–C所示)等表型在DKO小鼠明显减轻(如图5A–C所示)。图D-E显示了NG2/CD31免疫荧光共染和定量分析结果。我们发现,与WT小鼠或Unc13b-/-小鼠相比,Ccm3-iecKO小鼠的小脑病变扩张血管周围弥漫着ANGPT2阳性的免疫荧光染色伴随着升高的TIE2蛋白磷酸化染色。在Ccm3-iecKO小鼠的病变小脑升高的TIE2蛋白磷酸化阳性染色能在DKO小鼠中被明显抑制(如图5F–G)。ELISA和Western blotting结果显示ANGPT2总蛋白在Ccm3-iecKO小鼠的小脑轻度升高但能被同时敲除Unc13b基因所抑制(如图5H–I所示)。我们在分离出来的Ccm3-iecKO小鼠的小脑的血管内皮细胞检测到显著升高的ANGPT2蛋白分泌,其在DKO小鼠中明显减少(如图5J所示)。这些结果强烈提示Unc13b基因敲除能通过抑制ECs的ANGPT2-TIE2信号通路,减轻因Ccm3基因敲除引起的神经血管的表型。
3.结论分析
本研究,我们报道血管内皮细胞特异性Ccm3基因敲除(Ccm3-iecKO)的小鼠产生的CCM损害表现为小脑出血、EC细胞连接的破坏、EC和PC的解离,这与小脑扩张的微血管周围升高的ANGPT2分泌有关。我们在Ccm3-iecKO小鼠小脑和视网膜组织发现升高的ANGPT2分泌,但未在肺组织和血样中发现升高的ANGPT2分泌。CCM3只在脑和视网膜对ANGPT2有作用,这为血管内皮细胞特异性Ccm3基因敲除的小鼠只在脑和视网膜组织产生CCM损害提供了一个可能的解释。体外研究证实血管内皮细胞Ccm3基因缺失能导致EC细胞连接的破坏、扩大的管腔形成和减少的周细胞募集。这些EC表型与Ccm3基因缺失内皮细胞中增高的ANGPT2分泌和释放有关,抑制胞吐作用或ANGPT2中和抗体能显著抑制Ccm3基因缺失的EC表型。这表明,CCM3能通过抑制胞吐作用介导的ANGPT2释放来维护正常的EC细胞连接、管腔形成和血管成熟。运用共聚焦和TIRF显微镜进行的机制研究表明,CCM3通过抑制UNC13B/VAMP依赖的胞吐作用,调控ANGPT2从脑血管内皮细胞的Weibel-Palade小体释放。运用遗传学工具,我们发现Unc13b基因敲除能抑制EC特异性Ccm3基因敲除引起的细胞连接的破坏和血管的扩大。阻断CCM损害的发展。更为重要的是,我们的结果证实了ANGPT2中和抗体在小鼠CCM模型能抑制CCM损害的进展,这与它能稳定EC细胞连接、EC-PC相互作用和血管完整性有关。我们的研究揭示了Ccm3基因突变导致CCM疾病发展的新机制,这为目前这种不可治愈疾病提供了一种新的治疗手段。
我们最近证实CCM3与GCKIII激酶复合物结合到UNC13的C2B结构域,从而抑制UNC13跟胞膜脂质的结合,这一步是胞吐作用需要的。本研究,我们提供了强有力的证据证实了CCM3通过UNC13B/VAMP依赖的胞吐作用,控制内皮细胞ANGPT2的分泌。1)我们发现UNC13B是表达在血管内皮细胞的主要亚型。并且,在血管内皮细胞UNC13B与CCM3及GCKIII家族成员STK24形成复合物。2)通过共聚焦荧光显微镜分析发现,在脑ECs中CCM3与VAMP3和VAMP8共定位。3)通过实时动态TIRF荧光显微镜分析发现,CCM3缺失的ECs胞吐作用增强,这可被同时沉默UNC13B表达所抑制。4)在脑ECs敲低CCM3(而不是CCM1或CCM2)能明显增强细胞ANGPT2的分泌,该作用能被同时沉默UNC13B或VAMP3所阻断。然而,ANGPT2的mRNA水平和ANGPT2的总蛋白水平在CCM3沉默的脑ECs中不受改变。并且,ANGPT2蛋白稳定性和分泌在CCM3沉默的脑ECs中也未受改变。这些结果支持如下机制:因CCM3缺失引起的ANGPT2释放的增加来源于胞吐作用,而不是蛋白质合成或蛋白质稳定性。5)通过Ccm3和Unc13b基因敲除小鼠的遗传学手段,我们在小鼠脑和视网膜组织也发现CCM3-UNC13B调控ANGPT2的分泌。6)更重要的是,我们发现ANGPT2中和抗体能使EC连接正常化,并能在小鼠CCM模型中减缓CCM病变的进展。虽然Angpt2基因缺陷小鼠是可存活的,ANGPT2在出生后的血管生成和淋巴管形成过程中是必需的。ANGPT2缺陷能导致青光眼,一种致盲性疾病。临床上,有必要寻找最适剂量的ANGPT2,以期能达到最大程度的治疗CCM的效果和对生理性血管发育和功能有最小的副作用。
TIE2信号通路在血管静息和血管生成过程中起不同的作用。这归因于ANGPT1组装不同的TIE2信号复合物,在静息状态磷酸化的TIE2位于EC-EC相互接触处,而在血管生成过程中磷酸化的TIE2处于EC与基质接触处。尽管在静息状态下的内皮细胞ANGPT2对ANGPT1/TIE2信号起负调控作用。而在应激条件下的ECs,ANGPT2能激活TIE2磷酸化。我们在CCM3缺失的ECs发现增强的ANGPT2-TIE2信号,与之前的研究报道一致,即CCM3在ECs敲除能诱导几种应激相关的信号。我们的结果表明在CCM3缺失的脑组织和脑ECs中增强的ANGPT2分泌与升高的磷酸化的TIE2有关。在静息状态下的内皮细胞ANGPT2拮抗ANGPT1从而抑制TIE2的激活;而在血管生成过程中,它能协同ANGPT1促进TIE2的激活。我们的结果明确表明升高的ANGPT2-TIE2信号有助于CCM病理过程。然而,病理条件下由CCM3功能缺失引起ANGPT2-TIE2信号增强的后果跟急性炎症条件下ANGPT2-TIE2的激活在本质上可能不一样,因为只有CCM3功能缺失能快速地增大血管内皮管腔的膨胀。据报道,CCM1或CCM2功能缺失能在ECs激活几条信号通路,包括RhoA,MEKK3–ERK5–Krüppel-like factor(KLF)2/4信号和KLF4–TGF-β/Smad介导的内皮细胞–间充质转化的信号(endothelial-mesenchymaltransition,EndMT)。是否CCM3–UNC13–ANGPT2–TIE2信号轴能与上述信号通路存在相互作用,这需要进一步阐明。甚有意思的是,ANGPT1/TIE2能通过PI3K-Akt的激活,诱导静息血管内皮细胞中KLF2的表达。在CCM3功能缺失的ECs,ANGPT2通过TIE2诱导KLF2的表达,这可能是在CCM发展过程中CCM3介导ANGPT2–TIE2信号和CCM1/2介导MEKK3–ERK5信号的汇合点。
日渐清晰的是,受CCM三种蛋白调控的各个信号通路的一个共同的机制是它们都调控EC细胞连接,这揭示了为什么人类任何一个CCM基因突变或小鼠任何一个CCM基因敲除能导致类似的CCM血管病变的潜在的可能机制。CCM1与CCM2形成复合物,结合并调控影响EC细胞连接及其相关复合物的蛋白,如Rap1,HEG(heart of glass)和ICAP-1(integrin cytoplasmic domain associated protein-1)。我们的体外和体内实验结果明确表明CCM3通过抑制ANGPT2的分泌,调控脑ECs的粘附连接和紧密连接。ECs连接的改变如何与CCM损害发展相关,这未被明确阐明。CCM基因敲除的一个重要的和明确的表型是导致扩大的管腔形成。众所周知。ECs细胞连接的排列和重塑直接调控EC管腔形成。在小鼠胚胎早期发育过程中,编码基因VE-cadherin(Cdh5)纯合子突变尽管能引起主动脉和主静脉管腔的缩小,还能导致头静脉管腔的扩大。机制研究表明CCM1-VE-cadherin指引细胞粘附连接的组合排列,其极性复合物调控血管管腔结构,该理论也适用于CCM3。此外,CCM3介导的胞吐作用可能直接有助于管腔形成。现在普遍认为管腔形成是靠囊泡运输机制,与质膜的结构性扩张有关。最近有关果蝇气管管腔形成的研究报告也支持该理论。CCM3或GCKIII激酶同源物功能缺失的果蝇表现为扩张的气管形成,这与CCM病人扩张的血管相似。这种官腔扩张的表型能被低表达NSF2所抑制,NSF2是参与SNARE回收和胞吐作用的蛋白。我们知道,CCM病变好发于脑和视网膜血管,这与它们具有非常高的PC/EC比率,及有星形胶质细胞足突支撑的独特的血脑屏障和血-视网膜有关。我们的体外细胞模型研究也表明在内皮细胞CCM3功能缺失能导致管腔形成扩大和周细胞募集受损,这很好地模拟了动物模型中CCM病变的发展。而且,ANGPT2在调控CCM3介导的EC-PC相互作用方面起关键作用。因此,ANGPT2作为CCM3(而不是CCM1或CCM2)的下游效应因子,应该能解释为什么人类CCM3基因突变常导致更严重的疾病形态。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!