查耳烷类化合物及其制备方法和医药用途与流程
本发明属于医药技术领域,具体涉及疏叶崖豆叶子中的查耳烷类化合物及其制备方法和应用。
背景技术:
疏叶崖豆(Millettia pulchra Kurz var-laxior(Dunn)Z.Wei)是豆科(Leguminosae)崖豆藤属(Millettia)植物。崖豆藤属植物在我国有40种,主要分布于广西、广东、云南等地。
疏叶崖豆的干燥根又称为玉郎伞、龙眼参等,壮语称其为“捧吞”(广西靖西)、“肥药”(那坡)等;瑶医称其为“莽台”(人薯)、“木茯苓”(广西金秀)、“土茯神”(广西巴马)。具有补虚润肺、舒筋活络的功能,民间常用于高血压、跌打损伤、腰腿痛、风湿痛、慢性肝炎和结核的治疗,也可用于消化不良、营养不良和病后虚弱,还可以用于治疗再生障碍性贫血等。此外,本品还具有补气、补血、提高免疫力和改善大脑学习记忆等功效。现代药理研究表明该属植物具有抗肿瘤、保肝和抗雌性激素等多种药理活性。
査耳酮类天然产物在植物中分布广泛,具有显著的抗癌、抗炎、抗氧化、抗菌等生物活性。同时査耳酮类化合物是天然的迈克尔反应受体分子,参与体内多个代谢反应,对二相代谢酶的表达具有重要调控作用,是潜在的天然来源癌症化学预防剂。前期研究中我们发现查耳酮类化合物是疏叶崖豆地上部分的主要化学成分,结构类型丰富、含量高、且具有显著的体外癌症预防活性。本发明中描述了5个新查耳烷类化合物,具有显著的醌还原酶诱导活性,有可能用于制备预防和治疗癌症的药物。
技术实现要素:
本发明的目的在于提供一系列新的查耳烷及其制备方法和医药用途。具体提供了5个新的查耳烷millpuline B(1)、millpuline C(2)、millpuline D(3)、millpuline E(4)和millpuline F(5);
本发明所提供的查耳烷化合物的结构通式为:
(I)
其中,R1=H、OH、C1-C4烷氧基或OCnH2nOH(n=1-4);
R2=H、OH、C1-C4烷氧基或OCnH2nOH(n=1-4);
R3=H、OH、C1-C4烷氧基或OCnH2nOH(n=1-4);
R4=H、OH、C1-C4烷氧基;
R5=H、OH、C1-C4烷氧基。
本发明具体公开了如下五个具体化合物:
本发明还提供了所述新的查耳烷1~5的制备方法,该方法包括如下步骤:
(1)疏叶崖豆(Millettia pulchra Kurz var-laxior(Dunn)Z.Wei)干燥地上部分用50%~95%乙醇提取,回收提取液得粗提物;
(2)步骤(1)所得粗提物经大孔吸附树脂色谱法分离,以体积比为30:70~90:10乙醇-水或甲醇-水的混合溶剂梯度洗脱,得到不同极性部位的醇洗脱物;
(3)上述步骤(2)中所得的洗脱物经硅胶柱色谱,用石油醚/乙酸乙酯,石油醚/丙酮,二氯甲烷/丙酮,二氯甲烷/甲醇,氯仿/丙酮或氯仿/甲醇的混合溶剂梯度洗脱;
(4)上述步骤(3)中所得的流分经ODS柱色谱,用甲醇-水混合溶剂或乙腈-水混合溶剂梯度洗脱;
(5)上述步骤(4)所得流分,经HPLC-UV色谱分离,以甲醇-水混合溶剂,或乙腈-水溶剂为流动相洗脱,得到查耳烷1~5;
本发明提供的新的查耳烷1~5的制备方法,所述的疏叶崖豆叶子为豆科(Leguminosae)崖豆藤属(Millettia)疏叶崖豆(Millettia pulchra Kurz var-laxior(Dunn)Z.Wei)的干燥地上部分。
本发明提供的所述的新的查耳烷1~5的制备方法,步骤(1)中所述的提取方法为加热回流或加热超声提取1~3次。所用溶剂为50%~95%乙醇,优选70%~95%乙醇。药材:溶剂的重量体积为1:8~1:20,优选1:10~1:15。
本发明提供的所述新的查耳烷1~5的制备方法,步骤(2)中所述乙醇-水混合溶剂,或甲醇-水的混合溶剂中,混合溶剂的比例为甲醇:水或乙醇:水为30:70~90:0。
本发明提供的所述新的查耳烷1~5的制备方法,上述步骤(2)中所述的大孔树脂吸附法,采用丙酮溶解拌样的方法,将粗提物溶解于丙酮中(粗提物与丙酮的重量体积比W/V,1:1~1:6,优选为1:2~1:4)得该粗提物的丙酮溶液,加入1:2~1:10(优选1:3~1:5)倍量的大孔树脂搅拌均匀,减压浓缩至干。
本发明提供的所述新的查耳烷1~5的制备方法,步骤(3)中所述的石油醚/乙酸乙酯,石油醚/丙酮的混合溶剂体积比例为100:0~1:1,优选100:2~100:10;二氯甲烷/丙酮,氯仿/丙酮的混合溶剂的体积比例为100:0~2:1,优选100:1~100:8;二氯甲烷/甲醇,氯仿/甲醇的混合溶剂的体积比例为100:0~5:2,优选100:1~100:5。
本发明提供的所述新的查耳烷1~5的制备方法,步骤(4)中所述的甲醇-水的混合溶剂体积比例为30:70~100:0,优选50:50~90:10,乙腈-水的混合溶剂的体积比例为10:90~70:30,优选20:80~60:40。
本发明提供的所述新的查耳烷1~5的制备方法,步骤(5)中所述甲醇-水的混合溶剂的体积比例为50:50~95:5,优选70:30~90:10,乙腈-水的混合溶剂的体积比例为30:70~60:40,优选35:75~45:55。
本发明以体外Hepa 1c1c7细胞进行了醌还原酶[NQO1,NAD(P)H quinine oxidoreductase 1]诱导活性测试,对制备得到的新查耳烷1~5的NQO1活性进行 了评价。结果显示,这些新查耳烷具有显著NQO1诱导活性,可用于开发癌症化学预防剂或癌症治疗药物。
本发明首次提供了以疏叶崖豆地上部分为原料,制备、鉴定5个新查耳烷的方法,并且系统评价了NQO1诱导方面的活性,阐述了其在开发癌症化学预防、治疗药物方面的应用。
附图说明
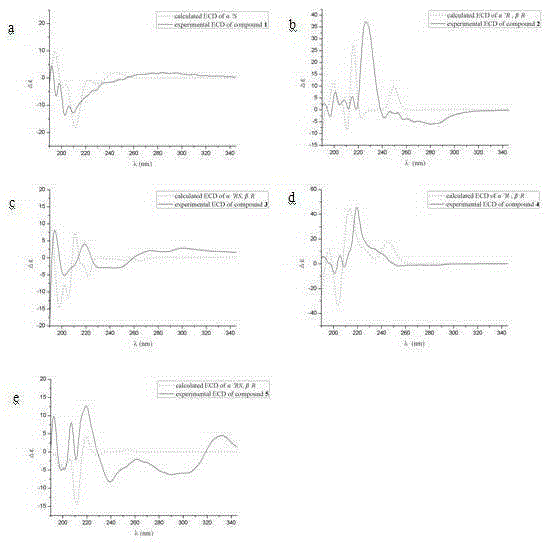
图1查耳烷1-5的ECD谱;
a.为查尔烷1的ECD谱;b.为查尔烷2的ECD谱;c.为查尔烷3的ECD谱;
d.为查尔烷4的ECD谱;e.为查尔烷5的ECD谱
图2查耳烷1的1H NMR谱;
图3查耳烷1的13C NMR谱;
图4查耳烷1的HMBC谱;
图5查耳烷2的1H NMR谱;
图6查耳烷2的13C NMR谱;
图7查耳烷2的HMBC谱;
图8查耳烷3的1H NMR谱;
图9查耳烷3的13C NMR谱;
图10查耳烷3的HMBC谱;
图11查耳烷4的1H NMR谱;
图12查耳烷4的13C NMR谱;
图13查耳烷4的HMBC谱;
图14查耳烷5的1H NMR谱;
图15查耳烷5的13C NMR谱;
图16查耳烷5的HMBC谱。
具体实施方式
下面的实施例将对本发明予以进一步的说明,但并不因此限制本发明。
实施例1
(1)疏叶崖豆干燥地上部分500g用70%乙醇提取1次(用量为10L),减压回收提取液的粗提物;
(2)上述步骤(1)所得70%乙醇粗提物经大孔树脂吸附(粗提物:丙酮=1:1,粗提物:大孔树脂=1:3),用水、30%乙醇、60%乙醇和90%乙醇梯度洗脱,得到不同极性部位的洗脱物;
(3)步骤(2)中90%乙醇洗脱物,经硅胶柱色谱分离,依次以石油醚和乙酸乙酯混合溶剂100:0,100:3,100:8,100:10洗脱;
(4)上述步骤(3)中所得的石油醚:乙酸乙酯100:3~100:8流分经ODS色谱,用30:70,50:50,70:30,90:10甲醇-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得甲醇-水(50:50~90:10)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为甲醇:水=78:22,得到查耳烷1(tR=110min)(收率为0.00033%),得到查耳烷2(tR=84min)(收率为0.0017%),得到查耳烷3(tR=94min)(收率为0.0035%)。
(6)上述步骤(4)中所得甲醇-水(50:50~90:10)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以70:30甲醇-水的混合溶剂为流动相,得到查耳烷4(tR=95min)(收率为0.0004%)和5(tR=64min)(收率为0.0003%)。
根据查耳烷1~5的理化性质和波谱数据鉴定了其结构(查耳烷1~5的ECD和HMBC、HMQC及其1H-1HCOSY相关图见附图1-10)。
查耳烷1的结构鉴定数据如下:
黄色油状物(MeOH),HR-ESI-MS给出准分子离子峰m/z 319.1297[M+Na]+:(calcd.for 319.1305,C19H20O3Na),得其分子式为C19H20O3,不饱和度为10。1H NMR和13C NMR数据见表1和2。HMBC和1H-1HCOSY相关信号见图8。该化合物的绝对构型通过运用TDDFT(时间依赖的密度泛函理论)方法进行ECD计算确定,将实验测出的谱图和计算得到的α′R的ECD图谱进行比较,图谱相吻合,表现在在190-195nm区域有正Cotton效应,在200-240nm区域有负Cotton效应(见附图7)。因此,确定该化合物的绝对构型为α′R。
查耳烷2的结构鉴定数据如下:
黄色油状物(MeOH),HR-ESI-MS给出准分子离子峰m/z 349.1414[M+Na]+:(calcd for 349.1410,C20H22O4Na),给出分子式为C20H22O4,不饱和度为10。采用与1相同的方法确定其绝对构型,2的ECD实 测值与差向异构体(α′R,βR)的计算值ECD图谱相吻合,表现在210-220nm和220-240nm区域有正Cotton效应,所以确定化合物的绝对构型为(α′R,βR)。
查耳烷3的结构鉴定数据如下:
黄色油状物(MeOH),HR-ESI-MS给出准分子离子峰m/z 349.1415[M+Na]+:(calcd for 349.1410,C20H22O4Na),给出分子式为C20H22O4,不饱和度为10。该化合物的绝对构型采用与查耳烷1相同的ECD计算方法,查耳烷3的ECD实测值与(α′S,βR)的计算值ECD图谱相吻合,表现为200-220nm区域有负Cotton效应,225-235nm区域有正Cotton效应,确定查耳烷3的绝对构型为(α′S,βR)。
查耳烷4的结构鉴定数据如下:
黄色油状物(MeOH),HR-ESI-MS给出准分子离子峰m/z 379.1503[M+Na]+(cacl for 379.1516,C21H24O5Na),推测出分子式为C21H24O5,不饱和度为10。该化合物的绝对构型采用与查耳烷1相同的ECD计算方法,查耳烷4的ECD实测值与(α′R,βR)的计算值ECD图谱相吻合,表现为180-190nm和195-205nm区域有负Cotton效应,220-250nm区域有正Cotton效应。因此确定查耳烷4的绝对构型为(α′R,βR)。
查耳烷5的结构鉴定数据如下:
黄色油状物(MeOH),HR-ESI-MS给出准分子离子峰m/z 379.1517[M+Na]+(cacl for 379.1516),推测出分子式为C21H24O5Na,不饱和度为10。该化合物的绝对构型采用与查耳烷1相同的ECD计算方法,查耳烷5的ECD实测值与(α′S,βR)的计算值ECD图谱相吻合,表现为205-215nm和220-230nm区域有正Cotton效应,180-190nm和195-205nm区域有负Cotton效应,所以查耳烷5的绝对构型为(α′S,βR)。
查耳烷1~5的NMR数据归属见表1和2
表1查耳烷1~5的1H NMR(400MHz,CDCl3)数据
表2查耳烷1~5的13C NMR(100MHz,CDCl3)数据
实施例2
(1)疏叶崖豆叶子1000g用95%乙醇提取3次(用量为8L),减压回收提取液的粗提物;
(2)上述步骤(1)中所得乙醇粗提物经大孔树脂吸附(丙酮:粗提物=1:2,粗提物:大孔树脂=1:5),用50%乙醇和90%乙醇梯度洗脱,得到不同极性部位的洗脱物;
(3)上述步骤(2)中90%乙醇洗脱物,经硅胶柱色谱分离,依次以石油醚和丙酮混合溶剂100:2,100:3,100:5,100:8,100:10洗脱;
(4)上述步骤(2)中所得的石油醚:丙酮100:2~100:10流分经ODS色谱,用10:90,30:70,50:50,70:30乙腈-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得乙腈-水(50:50)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为乙腈:水=40:60,得到查耳烷1(tR=100min)(收率为0.00038%),得到查耳烷2(tR=75min)(收率为0.0019%),得到查耳烷3(tR=89min)(收率为0.0037%)。
(6)上述步骤(4)中所得乙腈-水(70:30)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以35:65乙腈-水的混合溶剂为流动相,得到查耳烷4(tR=90min)(收率为0.0004%)和5(tR=59min)(收率为0.0003%)。实施例3
(1)疏叶崖豆叶子800g用50%乙醇提取2次(用量为9.6L),减压回收提取液的粗提物;
(2)上述步骤(1)所得乙醇粗提物经大孔树脂吸附(粗提物:丙酮=1:6,粗提物:大孔树脂=1:10),用30%乙醇、50%乙醇和80%乙醇梯度洗脱,得到不同极性部位的洗脱物;
(3)步骤(2)中80%乙醇洗脱物,经硅胶柱色谱分离,依次以二氯甲烷:丙酮混合溶剂100:1,100:3,100:5,100:8洗脱;
(4)上述步骤(3)中所得的二氯甲烷:丙酮100:3~100:8流分经ODS色谱,用50:50,70:30,90:10甲醇-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得甲醇-水(90:10)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为甲醇:水=82:18,得到查耳烷1 (tR=101min)(收率为0.00035%),得到查耳烷2(tR=76min)(收率为0.0014%),得到查耳烷3(tR=93min)(收率为0.0032%)。
(6)上述步骤(4)中所得甲醇-水(70:30)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以甲醇-水(77:23)的混合溶剂为流动相,得到查耳烷4(tR=65min)(收率为0.00037%)和5(tR=50min)(收率为0.00029%)。实施例4
(1)疏叶崖豆叶子1200g用75%乙醇提取2次(用量为18L),减压回收提取液的粗提物;
(2)上述步骤(1)所得乙醇粗提物经大孔树脂吸附(粗提物:丙酮=1:4,粗提物:大孔树脂=1:2),用35%和75%乙醇-水的混合溶剂梯度洗脱,得到不同极性部位的乙醇洗脱物;
(3)步骤(2)中75%乙醇洗脱物,经硅胶柱色谱分离,依次以二氯甲烷/甲醇混合溶剂100:1,100:3,100:5,100:8,100:10,3:1洗脱;
(4)上述步骤(2)中所得的二氯甲烷:甲醇100:1~100:5流分经ODS色谱,用30:70,50:50,90:10甲醇-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得甲醇-水(90:10)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为乙腈:水=42:58,得到查耳烷1(tR=92min)(收率为0.00030%),得到查尔烷2(tR=69min)(收率为0.0020%),得到查耳烷3(tR=80min)(收率为0.0031%)。
(6)上述步骤(4)中所得甲醇-水(50:50)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以36:64乙腈-水的混合溶剂为流动相,得到查耳烷4(tR=85min)(收率为0.00038%)和5(tR=55min)(收率为0.00027%)。实施例5
(1)疏叶崖豆叶子600g用80%乙醇提取3次(用量为10L),减压回收提取液的粗提物;
(2)上述步骤(1)所得乙醇粗提物经大孔树脂吸附(粗提物:丙酮=1:5,粗提物:大孔树脂=1:10),用30%、60%和90%甲醇-水的混合溶剂梯度洗脱,得到不同极性部位的甲醇洗脱物;
(3)步骤(2)中90%甲醇洗脱物,经硅胶柱色谱分离,依次以氯仿/丙酮混合溶剂100:1,100:2,100:3,100:5,100:6,100:8,100:10,3:1梯度洗脱;
(4)上述步骤(2)中所得的氯仿:丙酮100:3~100:8流分经ODS色谱,用5:95,15:85,25:75,35:65,40:60,45:55,60:40乙腈-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得乙腈-水(40:60)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为乙腈:水=45:55,得到查耳烷1(tR=89min)(收率为0.00029%),得到查尔烷2(tR=65min)(收率为0.0025%),得到查耳烷3(tR=75min)(收率为0.0030%)。
(6)上述步骤(4)中所得乙腈-水(35:65)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以40:60乙腈-水的混合溶剂为流动相,得到查耳烷4(tR=75min)(收率为0.00035%)和5(tR=48min)(收率为0.00024%)。实施例6
(1)疏叶崖豆叶子300g用,65%乙醇提取2次(用量为3L),减压回收提取液的粗提物;
(2)上述步骤(1)所得乙醇粗提物经大孔树脂吸附(粗提物:丙酮=1:4,粗提物:大孔树脂=1:8),用30%、50%和70%和90%乙醇-水的混合溶剂梯度洗脱,得到不同极性部位的乙醇洗脱物;
(3)步骤(2)中90%乙醇洗脱物,经硅胶柱色谱,依次以氯仿/甲醇混合溶剂100:0,100:1,100:3,100:5,100:10,5:2梯度洗脱;
(4)上述步骤(2)中所得的氯仿/甲醇100:1~100:5流分经ODS色谱,用20:80,30:70,50:50,60:40乙腈-水的混合溶剂梯度洗脱;
(5)上述步骤(4)中所得乙腈-水(60:40)流分经HPLC-UV色谱分离制备,210nm检测,流速为4mL/min,流动相为甲醇:水=90:10,得到查耳烷1(tR=88min)(收率为0.00029%),得到查尔烷2(tR=63min)(收率为0.0025%),得到查耳烷3(tR=72min)(收率为0.0030%)。
(6)上述步骤(4)中所得乙腈-水(60:40)流分经HPLC-UV色谱分离,210nm检测,流速为4mL/min,以72:28甲醇-水的混合溶剂为流动相,得到查耳烷4(tR=88min)(收率为0.00035%)和5(tR=57min)(收率为0.00024%)。 实施例7实施例1中制备所得新查耳烷1-5的醌还原酶诱导活性研究
(1)实验原理
鼠肝癌细胞系Hepa lclc7细胞可用于简单测NQO1的诱导活性。当存在NADP时,葡萄糖-6-磷酸可以被葡萄糖-6-磷酸脱氢酶分解,产生NADPH,其可以作为电子供体,使甲萘醌变为甲萘氢醌。甲萘氢醌可以使MTT转变为甲臜(formazan),在550nm下用于定量测定。
(2)实验方法
将Hepa lclc7细胞养于96孔板中,当细胞长满80%时给药。给药培养24h后,弃去培养基,每孔加入50μL细胞裂解液(0.8%w/v洋地黄皂苷,含2mM EDTA)。振摇裂解10min后加入200μL现配的“完全反应液”(40mL反应液组成:2mL0.5M Tris-HCL(PH 7.4),0.27mL1.5%Tween-20,0.027mL 7.5mLflavin adenine dinucletide,0.27mL150Mm 6-磷酸葡萄糖,0.024mL50mM NADP,26.7mg牛血清白蛋白,12mg的MTT,0.04mLof 50mM甲苯醌,80units葡萄糖-6-磷酸脱氢酶,加去离子水补充至40mL)。摇匀,5min后,酶标仪550nm下测定吸光值。
将样品储备液溶于DMSO中,给药时DMSO浓度保持在0.5%(v/v)。4'-溴黄酮(终浓度4μM)与DMSO分别作为阳性与阴性对照,两者DMSO浓度均保持为0.5%(v/v)。计算IR值(相对于阴性水平的NQO1诱导倍数)与CD值(NQO1诱导活性二倍于阴性水平时的给药浓度)作为NQO1诱导活性。
(3)实验结果
实验结果见表3
表3化合物1~5的细胞毒活性和NQO1诱导活性(浓度10μM)
- 还没有人留言评论。精彩留言会获得点赞!