用于分析检测和筛选半乳糖激酶抑制剂的荧光探针的制作方法
本发明属于生物分析检测领域,特别涉及一种用于分析检测和筛选半乳糖激酶抑制剂的荧光探针及制备方法及应用。
技术背景
半乳糖血症是血和尿中半乳糖增多的一种疾病,新生儿或婴幼儿多出现的该疾病是由于半乳糖-l-磷酸尿苷酰转移酶(GALT)遗传性缺失,从而导致半乳糖的第一步代谢产物半乳糖-l-磷酸(Gal-1-P)在患者体内组织和细胞内的大量积蓄而造成的。半乳糖血症的主要症状是营养障碍、白内障、黄疸,腹泻,脓毒症,智力低下和肝脾肿大,新生儿死亡等。半乳糖血症的临床治疗方法主要是静脉葡萄糖补充疗法,新鲜血浆输注,电解质补充以及针对合并败血症患者进行抗生素治疗。
半乳糖在分子结构上和糖分类中与葡萄糖是截然不同的单糖。食物中的半乳糖主要来自奶类所含的乳糖。哺乳婴儿所需能量的20%由乳类中的乳糖提供。正常情况下,乳糖进入肠道后即被水解成半乳糖和葡萄糖经肠粘膜吸收。半乳糖被吸收后在细胞内先后经半乳糖激酶(GALK)、半乳糖-l-磷酸尿苷酰转移酶(GALT)和尿苷二磷酸半乳糖表异构酶(GALE)的作用,最终生成l-磷酸葡萄糖进入葡萄糖代谢途径。由于半乳糖的代谢产物半乳糖-l-磷酸(Gal-1-P)在细胞和体内的代谢物积蓄是导致各种脏器官器质性病变的主要原因,因此从根本上预防和治疗半乳糖血症的方法之一是通过有效抑制半乳糖代谢通路中的半乳糖激酶(GALK)避免半乳糖的代谢产物半乳糖-l-磷酸(Gal-1-P)在细胞内的积蓄,从而达到保持患者健康状态的目的(Liu L,Tang M,Walsh MJ,Brimacombe KR,Pragani R,Tanega C,Rohde JM,Baker HL,Fernandez E,Blackman B1,Bougie JM,Leister WH,Auld DS,Shen M,Lai K,Boxer MB,Bioorg Med Chem Lett.2015Feb 1;25(3):721-727)。因此,发现和开发能够有效抑制半乳糖激酶活性的GALK激酶抑制剂,对预防和治疗半乳糖血症非常重要。
研究表明,半乳糖激酶(GALK)对半乳糖的代谢过程首先是激酶分子中的天冬氨酸残基对半乳糖结构中的1-位羟基进行去离子化,然后从细胞中的ATP获取一分子磷酸从而完成半乳糖-l-磷酸(Gal-1-P)的生成(参考文献:Megarity CF,Huang M,Warnock C,Timson DJ.Bioorg Chem.2011Jun;39(3):120-6.)。上述代谢过程具有很高的特异性,原因是半乳糖激酶不仅特异性地识别半乳糖(而不是其他的单糖。比如葡萄糖,果糖,甘露糖等),而且半乳糖分子内的1-位羟基的存在对半乳糖成为半乳糖激酶的代谢底物亦非常重要。(见图9)
迄今应用于筛选和发现GALK抑制剂的方法,主要是利用半乳糖激酶将半乳糖转化为半乳糖-l-磷酸过程中需要定量消耗ATP的特点,需要利用在实验室制备及纯化后获得的半乳糖激酶蛋白,通过向纯化后的激酶蛋白中加入定量的半乳糖底物和定量的ATP,然后测试由于抑制剂的抑制作用而导致ATP消耗减少最终判断抑制剂的效果和强弱(文献:Liu L,Tang M,Walsh MJ,Brimacombe KR,Pragani R,Tanega C,Rohde JM,Baker HL,Fernandez E,Blackman B1,Bougie JM,Leister WH,Auld DS,Shen M,Lai K,Boxer MB,Bioorg Med Chem Lett.2015Feb 1;25(3):721-727)。这种筛选方法涉及到繁琐的半乳糖激酶的制备与纯化,而且抑制剂本身与ATP的相互作用会对测试结果产生不可避免的干扰。
技术实现要素:
本发明的目的是克服现有技术的不足,提供一种用于分析检测和筛选半乳糖激酶抑制剂的荧光探针。
本发明的第二个目的是提供一种用于分析检测和筛选半乳糖激酶抑制剂的荧光探针的制备方法。
本发明的第三个目的是提供一种用于分析检测和筛选半乳糖激酶抑制剂的荧光探针分析检测和筛选半乳糖激酶抑制剂中的用途。
本发明的技术方案概述如下:
一种用于分析检测和筛选半乳糖激酶抑制剂的荧光探针,其结构如式(I)所示:
其中,
X为O或NH;
R2较好地选自:(A-1)、(A-2)、(A-3)、(A-4)或(A-5);
n=0‐4。
最好是选自:
式中,n=1‐4。
R2还可以选自表1列举的具有不同激发波长的荧光团:
表1
用于分析检测和筛选半乳糖激酶抑制剂的荧光探针的制备方法,包括如下步骤:
(1)在室温条件下,将式(A-6)或式(A-7)所示的化合物与6-氨基-6-脱氧半乳糖化合物(A-8)进行缩合,得到式(A-9)或式(A-10)所示的半乳糖偶联酰胺产物;其中,X=O或NH;Y=Cl或OH;n=1-4;R3和R4相同或者不同,为羟基保护基;或者R3和R4分别代表邻位羟基的缩醛或缩酮保护基;
反应式如下:
(2)将步骤(1)获得的(A-9)或(A-10)中的羟基保护基脱保护,或者R3和R4代表的邻位羟基的缩醛或缩酮保护基脱保护,得到式(I)所示用于分析检测和筛选半乳糖激酶抑制剂的荧光探针,其中,X为O或NH;R2选自(A-4)或(A-5);n=1-4。
在缩合反应时使用多肽缩合试剂,所述多肽缩合试剂为N,N’-二环己基碳二亚(DCC),1-羟基苯并三唑(HOBt),六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(PyBOP)或O-苯并三氮唑-N,N,N,N-四甲基脲四氟硼酸酯(TBTU),O-苯并三氮唑-四甲基脲六氟磷酸盐(HBTU)。
在缩合反应时使用有机碱,所述有机碱为4-二甲氨基吡啶,三乙基胺或二异丙基乙胺。
羟基保护基为乙酰基或苄基。
邻位羟基的缩醛或缩酮保护基为1,2,3,4-二-氧-异丙叉基。
上述用于分析检测和筛选半乳糖激酶抑制剂的荧光探针分析检测和筛选半乳糖激酶抑制剂中的用途。
本发明具有以下的有益效果:
提供了一种能够直接用在细胞体系中分析、检测和筛选半乳糖激酶抑制剂的荧光探针,该探针能够简化以往的半乳糖激酶抑制剂筛选过程,避免使用提纯精制后的半乳糖激酶蛋白,而直接用于任何表达半乳糖激酶的细胞体系中;应用本发明的荧光探针能够方便快速地实现分析、测试和筛选半乳糖激酶抑制剂的目的,为发现和开发能够用于预防和治疗半乳糖血症疾病的先导化合物和有效药物提供了有效的手段和有用的工具。
附图说明
图1为本发明荧光探针化合物I-1的激发及发射荧光光谱;
图2为本发明荧光探针化合物I-3的激发及发射荧光光谱;
图3为本发明荧光探针化合物I-5的激发及发射荧光光谱;
图4为本发明荧光探针化合物I-7的激发及发射荧光光谱;
图5为本发明荧光探针化合物I-1在血红细胞中筛选半乳糖激酶抑制剂的实验结果;
图6为本发明荧光探针化合物I-3在血红细胞中筛选半乳糖激酶抑制剂的实验结果;
图7为本发明荧光探针化合物I-5在血红细胞中筛选半乳糖激酶抑制剂的实验结果;
图8为本发明荧光探针化合物I-7在血红细胞中筛选半乳糖激酶抑制剂的实验结果。
图9为半乳糖激酶特异性识别体内半乳糖并对其1-位羟基进行磷酸化代谢的示意图。
具体实施方式
设计理念是创造一种带有荧光的半乳糖激酶代谢底物(荧光探针),当荧光探针与细胞内半乳糖激酶的相互作用受到半乳糖激酶抑制剂的影响时产生荧光强度的变化,从而通过测试荧光探针因抑制剂的竞争性结合在细胞内产生的荧光信号变化,达到发现和筛选半乳糖激酶抑制剂的目的。
荧光探针含有半乳糖结构,并且在半乳糖6‐位连接荧光团,同时能够被半乳糖激酶识别,通过与半乳糖激酶抑制剂产生竞争从而用于分析、检测和筛选潜在的半乳糖激酶抑制剂。
本发明的用于分析检测和筛选半乳糖激酶抑制剂的荧光探针可经过下述合成路线制备获得:
方法A:
方法B:
方法A中,带荧光团的酰氯可以遵循文献的类似方法制备获得(Bioorganic and Medicinal Chemistry Letters,2008,vol.18,#3p.1106-1109)。在等当量的有机碱,例如4-二甲氨基吡啶,二乙胺,二异丙基乙胺或者吡啶的存在下带荧光团的酰氯与羟基保护的6-羟基或者6-氨基半乳糖,例如,商业购买的1,2:3,4-氧-异丙叉基-α-D-呋喃半乳糖(CAS号:4064-06-6),或者6-氨基-6-脱氧-1,2,3,4-二-氧-异丙叉基-α-D-半乳糖(CAS号:4711-01-7),在N,N-二甲基甲酰胺(DMF),二氯甲烷(DCM)等有机溶剂中室温反应便可制备获得荧光团与半乳糖偶联的中间产物,该中间产物经过脱去半乳糖羟基保护基,便可最后制得目标荧光探针。脱保护的条件据羟基保护基的种类而定,例如当使用酰基保护半乳糖羟基时,可以使用碳酸钾在甲醇和水的混合溶剂中脱保护,亦可以在室温使用氢氧化钠,氢氧化钾或者氢氧化锂在甲醇和水的混合溶剂中脱保护。
方法B中,带荧光团的羧酸原料可以遵循文献的方制备获得(法Bioorg Med Chem.2015Apr 15;23(8):1758-62.),该原料与羟基保护的6-羟基或者6-氨基半乳糖,例如,商业购买的1,2:3,4-氧-异丙叉基-α-D-呋喃半乳糖(CAS号:4064-06-6),或者6-氨基-6-脱氧-1,2,3,4-二-氧-异丙叉基-α-D-半乳糖(CAS号:4711-01-7)直接在肽合成反应常用的多肽缩合试剂,例如N,N’-二环己基碳二亚(DCC),1-羟基苯并三唑(HOBt),六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(PyBOP)或O-苯并三氮唑-N,N,N,N-四甲基脲四氟硼酸酯(TBTU),O-苯并三氮唑-四甲基脲六氟磷酸盐(HBTU)等的存在下进行缩合反应制备荧光团与半乳糖偶联的中间产物,所使用的溶剂可以是DMF,DCM,THF(四氢呋喃),NMP(N-甲基吡咯烷酮)以及它们的任意混合溶剂,反应温度一般为零度至60℃,缩合反应一般添加等当量的有机碱,例如4-二甲氨基吡啶,二乙胺,二异丙基乙胺等。第二步的脱保护反应,如果使用缩丙酮保护1,2-位和3,4-位的羟基,脱保护时可以使用三氟乙酸和少量的水在室温下完成。
下面以实施例进一步说明本发明,但不限制本发明。
实施例1:
化合物(I-1)按照以下方式合成
(1)化合物1a的合成:
1)在室温条件下将cy3(246.5mg)加入到干燥的DMF(二甲基甲酰胺)(10ml)当中,冷却到0℃,用氮气置换烧瓶内空气,在氮气保护下加入TBTU(O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯)(176.5mg)。
2)将反应液在氮气保护下室温搅拌15分钟,然后顺序加入6-氨基-6-脱氧-1,2,3,4-二-氧-异丙基-α-D-半乳糖(142.62mg)和三乙胺(TEA)(151.8mg),反应液在室温下反应12小时。反应完成后,旋蒸除去溶剂,二氯甲烷溶液萃取有机相,得粗产品使用硅胶柱色谱(二氯甲烷∶甲醇=45∶1)对反应生成物实施简单纯化,得到蓝色固体产品304.7mg。
收率:83.0%
1H NMR(600MHz,CDCl3)δ8.40(t,J=11.7Hz,1H),7.38(dd,J=21.4,6.2Hz,4H),7.25(d,J=7.3Hz,2H),7.13(d,J=7.7Hz,2H),6.75(dd,J=29.9,12.7Hz,2H),5.49(d,J=4.3Hz,1H),4.58(d,J=7.7Hz,1H),4.29–4.26(m,1H),4.22(d,J=7.7Hz,1H),4.10(s,2H),3.94(s,1H),3.72(s,3H),3.61–3.54(m,1H),3.30(s,1H),2.28(s,2H),1.85(s,2H),1.72(s,14H),1.60(s,2H),1.50(s,3H),1.43(s,3H),1.33(s,3H),1.30(s,3H).
(2)化合物I-1的合成:
将化合物1a(183.59mg)在10℃下溶解于三氟乙酸(TFA)-水(9ml:1ml)溶液当中,反应液在10℃下搅拌0.5小时,反应完成后,旋蒸除去溶剂,将残余物溶解于二氯甲烷溶液(70ml)和氯化铵溶液(70ml)中,水相用二氯甲烷(2x100)反萃2次,得有机相溶剂用无水硫酸钠干燥,减压旋干,硅胶色谱法(二氯甲烷:甲醇=10:1)纯化,得红色固体化合物(117.3mg)。
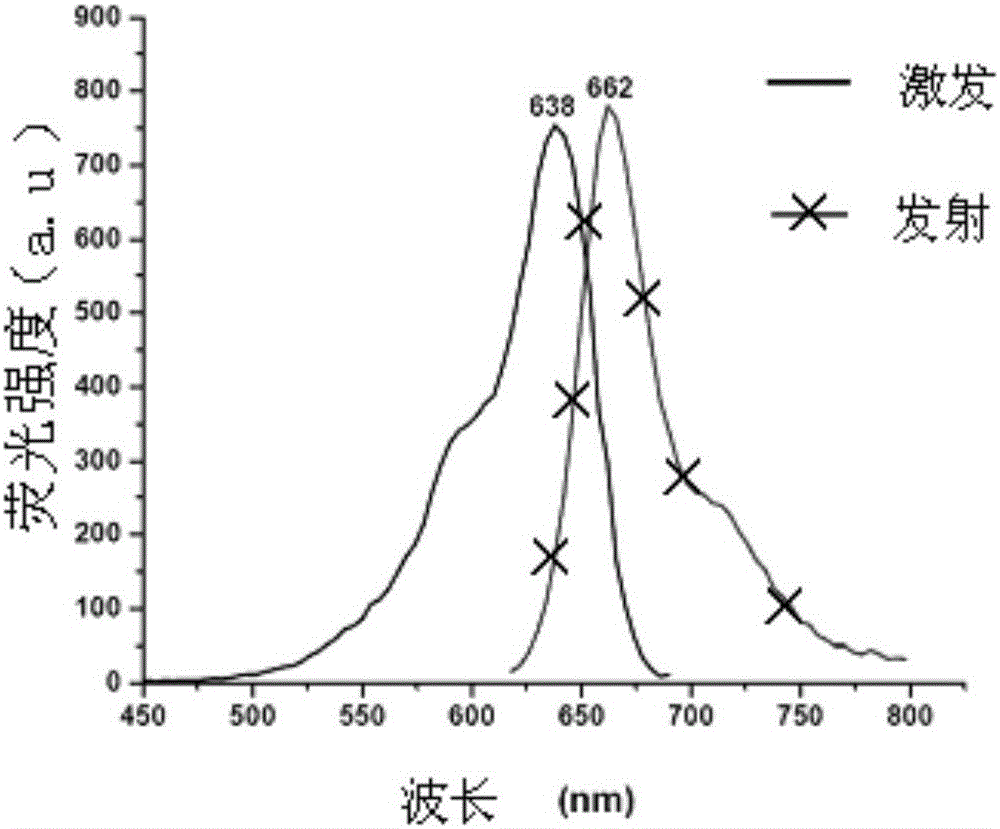
收率:71.7%(其激发和发射光谱见图-1)
1H NMR(600MHz,MeOD)δ8.45(t,J=13.4Hz,1H),7.44(d,J=7.4Hz,2H),7.35(t,J=7.6Hz,2H),7.24(dd,J=24.8,7.0Hz,4H),6.36(t,J=13.2Hz,2H),5.00(s)and 4.27(d,J=6.1Hz,1H),4.05(t,J=7.1Hz,2H),3.90(t,J=6.4Hz,1H),3.69–3.57(m,5H),3.39(ddd,J=33.7,23.1,6.3Hz,2H),3.12(dd,J=13.6,7.5Hz,1H),2.14(t,J=7.1Hz,2H),1.80–1.72(m,2H),1.72–1.52(m,14H),1.41(d,J=6.9Hz,2H).HRMS(ESI+):calculated for C36H48N3O6[M]+:618.3538;found:618.3538.
实施例2:
化合物(I-2)按照以下方式合成
(1)化合物2a的合成:
1)在室温条件下将cy3(200.5mg)和1,2:3,4-氧-异丙叉基-α-D-呋喃半乳糖(106.0mg)加入到干燥的DCM溶液(二氯甲烷)(10ml)当中,用氮气置换烧瓶内空气。
2)反应液中加入等当量4-二甲氨基吡啶(DMAP)然后在氮气保护下室温搅拌1小时。加入N,N’-二环己基碳二亚(N,N'-二环己基碳二亚胺)(100.8mg),反应液在室温下反应12小时。反应完成后,旋蒸除去溶剂,二氯甲烷溶液萃取有机相,得粗产品使用硅胶柱色谱(二氯甲烷∶甲醇=45∶1)对反应生成物实施简单纯化,得到红色固体产品133.1mg。
收率:44.5%
HRMS(ESI+):calculated for C42H55N2O7[M]+:699.4004;found:699.4001.
(2)化合物I-2的合成:
将化合物2a(133.1mg)在10℃下溶解于三氟乙酸-水(9ml:1ml)溶液当中,反应液在10℃下搅拌0.5小时,反应完成后,旋蒸除去溶剂,将残余物溶解于二氯甲烷溶液(70ml)和氯化铵溶液(70ml)中,水相用二氯甲烷(2x100)反萃2次,得有机相溶剂用无水硫酸钠干燥,减压旋干,硅胶色谱法(二氯甲烷:甲醇=10:1)纯化,得红色固体化合物(88.8mg)。收率:74.9%
HRMS(ESI+):calculated for C36H47N2O7[M]+:619.3378;found:619.3373.
实施例3:
化合物(I-3)按照以下方式合成
(1)化合物3a的合成:
参照化合物1a的合成方法,将起始原料改成cy5最后获得化合物3a。
收率:80.3%
1H NMR(600MHz,CDCl3)δ8.04(d,J=10.8Hz,2H),7.36(d,J=5.5Hz,4H),7.22(t,J=7.1Hz,2H),7.10(dd,J=15.2,7.8Hz,2H),6.70(s,1H),6.20(t,J=12.9Hz,2H),5.50(d,J=4.4Hz,1H),4.59(d,J=7.7Hz,1H),4.30(s,1H),4.22(d,J=7.7Hz,1H),4.00(s,2H),3.93(d,J=5.8Hz,1H),3.64(d,J=14.6Hz,4H),3.28(d,J=9.3Hz,1H),2.25(s,2H),1.81(s,2H),1.72(d,J=12.3Hz,14H),1.51(d,J=15.1Hz,2H),1.49(s,3H),1.45(s,3H),1.34(s,3H),1.31(s,3H).
(2)化合物I-3的合成::
参照I-1的合成方法,最后获得化合物I-3。
收率:68.1%(其激发和发射光谱见图-2)
1H NMR(600MHz,MeOD)δ8.26(t,J=12.9Hz,2H),7.51(d,J=7.3Hz,2H),7.43(t,J=7.6Hz,2H),7.30(dd,J=16.9,7.4Hz,4H),6.67(t,J=12.3Hz,1H),6.31(d,J=13.6Hz,2H),5.14(s)and 4.40(d,J=5.5Hz,1H),4.12(t,J=6.7Hz,2H),4.03(d,J=5.6Hz,1H),3.77(dd,J=24.8,12.3Hz,2H),3.65(s,3H),3.58–3.50(m,1H),3.46(s,1H),3.25(dd,J=13.6,7.5Hz,1H),2.26(t,J=7.0Hz,2H),1.89–1.82(m,2H),1.74(d,J=10.3Hz,16H).HRMS(ESI+):calculated for C38H50N3O6[M]+:644.3694;found:644.3708.
实施例4:
化合物(I-4)按照以下方式合成
(1)化合物4a的合成:
参照化合物2a的合成方法,将起始原料改成cy5最后获得化合物4a。
收率:42.9%
HRMS(ESI+):calculated for C44H57N2O7[M]+:725.4160;found:725.4153.
(2)化合物I-4的合成::
参照化合物I-2的合成方法,最后获得化合物I-4。
收率:69.1%
HRMS(ESI+):calculated for C38H49N2O7[M]+:645.3534;found:645.3531.
实施例5:
化合物(I-5)按照以下方式合成
(1)化合物5a的合成:
1)在室温条件下将荧光素羧酸原料(A-6-5)(165.2mg)加入到干燥的DMF溶液(10ml)当中,冷却到0℃,用氮气置换烧瓶内空气,在氮气保护下加入N,N’-二环己基碳二亚(85.3mg)和HOBT(57.6mg)。
2)将反应液在氮气保护下室温搅拌15分钟,然后顺序加入6-氨基-6-脱氧-1,2,3,4-二-氧-异丙基-α-D-半乳糖(87.5mg),反应液在室温下反应12小时。反应完成后,旋蒸除去溶剂,二氯甲烷溶液萃取有机相,得粗产品使用硅胶柱色谱(二氯甲烷∶甲醇=40∶1)对反应生成物实施简单纯化,得到蓝色固体产品164.2mg。收率:78.0%
(2)化合物I-5的合成::
将化合物5a(164.2mg)在10℃下溶解于三氟乙酸-水(9ml:1ml)溶液当中,反应液在10℃下搅拌0.5小时,反应完成后,旋蒸除去溶剂,将残余物溶解于二氯甲烷溶液(70ml)和氯化铵溶液(70ml)中,水相用二氯甲烷(2x100)反萃2次,得有机相溶剂用无水硫酸钠干燥,减压旋干,硅胶色谱法(二氯甲烷:甲醇=10:1)纯化,得红色固体化合物(109.8mg)。收率:75.6%(其激发和发射光谱见图-3)
1H NMR(400MHz,MeOD)δ8.45(td,J=13.5,1.5Hz,1H),7.48–7.17(m,8H),6.37(dd,J=
13.6,4.9Hz,2H),4.98(d,J=2.9Hz)and 4.13(d,J=7.6Hz,1H),4.33(m,2H),3.85(t,J=6.8Hz)and 3.50(d,J=3.0Hz,1H),3.62(d,J=1.8Hz,3H),3.56(d,J=4.1Hz,1H),3.40–3.23(m,2H),3.20–3.08(m,2H),2.63(t,J=6.5Hz,2H),1.68(d,J=1.1Hz,12H).HRMS(ESI+):calculated
for C33H42N3O6[M]+:576.3068;found:576.3068.
实施例6:
化合物(I-6)按照以下方式合成
(3)化合物6a的合成:
1)在室温条件下将荧光素羧酸原料(A-6-5)(269.5mg)和1,2:3,4-氧-异丙叉基-α-D-呋喃半乳糖(155.62mg)加入到干燥的DCM溶液(10ml)当中,用氮气置换烧瓶内空气。
2)在氮气保护下向反应液中加入等当量的4-二甲氨基吡啶于室温搅拌1小时,然后加入N,N’-二环己基碳二亚(148.0mg),反应液在室温下反应12小时。反应完成后,旋蒸除去溶剂,二氯甲烷溶液萃取有机相,得粗产品使用硅胶柱色谱(二氯甲烷∶甲醇=45∶1)对反应生成物实施简单纯化,得到红色固体产品190.7mg。
收率:45.9%
HRMS(ESI+):calculated for C39H49N2O7[M]+:657.3534;found:657.3530.
(4)化合物I-6的合成::
将化合物6a(190.7mg)在10℃下溶解于三氟乙酸-水(9ml:1ml)溶液当中,反应液在10℃下搅拌0.5小时,反应完成后,旋蒸除去溶剂,将残余物溶解于二氯甲烷溶液(70ml)和氯化铵溶液(70ml)中,水相用二氯甲烷(2x100)反萃2次,得有机相溶剂用无水硫酸钠干燥,减压旋干,硅胶色谱法(二氯甲烷:甲醇=10:1)纯化,得红色固体化合物(122.4mg)。收率:72.6%
HRMS(ESI+):calculated for C33H41N2O7[M]+:577.2908;found:577.2905.
实施例7:
化合物(I-7)按照以下方式合成
(1)化合物7a的合成:
参照化合物5a的合成方法,将起始原料改成相应的荧光素羧酸原料(A-7-7)羧酸化合物最后获得化合物7a。
收率:73.4%
(2)化合物I-7的合成::
参照化合物I-5的合成方法,最后获得化合物I-7。
收率:71.6%(其激发和发射光谱见图-4)
1H NMR(400MHz,MeOD)δ8.16(m,2H),7.27(m,8H),6.54(t,J=10.9Hz,1H),6.22(dd,J=26.6,13.6Hz,2H),5.00(s)and 4.16(d,J=7.6Hz,1H),4.28(d,J=3.6Hz,2H),3.87(m,1H),3.74–3.44(m,5H),3.43–3.24(m,2H),3.15(m,1H),2.72–2.53(m,2H),1.64(s,12H).HRMS(ESI+):calculated for C35H44N3O6[M]+:602.3225;found:602.3225.
实施例8:
化合物(I-8)按照以下方式合成
(3)化合物8a的合成:
参照化合物6a的合成方法,将起始原料改成荧光素羧酸原料(A-7-7)最后获得化合物8a。收率:49.8%
HRMS(ESI+):calculated for C41H51N2O7[M]+:683.3691;found:683.3683.
(4)化合物I-8的合成::
参照化合物I-6的合成方法,最后获得化合物8。
收率:70.6%
HRMS(ESI+):calculated for C35H43N2O7[M]+:603.3065;found:603.3060.
实验证明:用本发明上述实施例的方法可以制备式(I)所示的X为O或NH;R2为:(A-1)、(A-2)、(A-3)、(A-4)或(A-5);n=0‐4的化合物。
本发明还提供了所述荧光探针用于分析检测和筛选半乳糖激酶抑制剂中的用途。所发明的探针可用于任何表达半乳糖激酶蛋白的细胞体系,通过使用1-100μM的本发明探针,在细胞培养基存在的条件下,与细胞培养10-120min,便能使含有半乳糖激酶的细胞产生荧光染色。遵循上述方法使用所发明的探针可以分析和鉴定特定的化合物是否为半乳糖激酶抑制剂,当所鉴定的化合物为半乳糖激酶抑制剂时,所发明的探针便会在细胞内形成游离聚集,使细胞内的探针荧光强度增强。同样的道理,应用本发明的荧光探针可以在细胞体系中实施半乳糖激酶抑制剂的低通量或者高通量筛选。检测探针荧光强度的方法可以是荧光分光光度计,荧光酶标仪,荧光显微镜以及共聚焦显微镜等,所使用的激发波长根据具体的探针而定最好为500-650nm,本发明探针的发射波长范围为550-700nm。具体探针的激发和发射波长如附图所示。
试验例1:本发明荧光探针荧光光谱的测定实验
将探针化合物用DMSO溶解,PBS缓冲液稀释成50μM,用Thermo Scientific Varioskan LUX荧光酶标仪测定探针化合物的荧光光谱,以确定本发明探针化合物的荧光激发和发射波长。
本发明所提供的探针化合物分为Cy3系列和Cy5系列,其中,Cy3系列的探针化合物I-1,I-2,I-5,I-6的激发波长为:540nm至545nm,发射波长分别为:560nm和566nm;Cy5系列的探针化合物I-3,I-4,I-7,I-8的激发波长为:635至639nm,发射波长为660和666nm。
试验例2:本发明的荧光探针在筛选半乳糖激酶抑制剂中的应用
细胞培养(红细胞):
1.从血库购得过期的肝素化的人血液,2000g离心5分钟,吸去上层血浆和白细胞层。
2.用预冷的PBS(pH 7.4)重悬,吹吸10次,2000g离心5分钟;重复上述过程洗三遍,每次加入的体积为原来的3倍,直至上清清澈。
3.2000g离心5分钟,预冷的PBS重悬,配制成体积分数为5%的细胞悬液。四度保存,两周内使用。
抑制剂筛选:
实验测试用已知的半乳糖激酶抑制剂β-lapachone由Sigma-Aldrich购得。抑制剂用DMSO溶解,而后用PBS稀释到最终浓度。
红细胞先用DMEM培养30min,再无糖无血清1640饥饿30min,而后将抑制剂加入红细胞,培养2h,而后直接将本发明荧光探针加入其中,培养2h。冰冷PBS洗3遍,转移到酶标板中测量荧光强度变化。探针化合物I-1,I-2,I-5,I-6的最终浓度为40μM,测试激发波长设为543nm,发射波长设为565nm;探针化合物I-3,I-4,I-7,I-8的最终浓度为20μM,测试激发波长设为638nm,发射波长设为665nm。
探针在细胞内的荧光强度随抑制剂的浓度变化测试结果如下:
表-2:
表2中结果显示,本发明荧光探针在细胞内的荧光强度随着半乳糖激酶抑制剂浓度的变化而发生线性变化。根据此原理,本发明探针可以在不纯化半乳糖激酶蛋白的情况下,直接在细胞体系中应用于筛选未知的半乳糖激酶抑制剂。
探针化合物I-1,I-3,I-5,I-7在血红细胞中的荧光强度随半乳糖激酶抑制剂浓度变化见图5,图6,图7和图8.
实验证明:式(I)所示的X为O或NH;R2为:(A-1)、(A-2)、(A-3)、(A-4)或(A-5);n=0‐4的化合物具有上述效果。
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明,对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种快速检测BCR/ABL基因融合的探针组、试剂盒及方法与流程
- 酪氨酸激酶抑制剂的制造方法与工艺
- 芳香族酰胺类衍生物、其制备方法及其在医药上的应用与制造工艺
- 包含5α‑还原酶抑制剂的药物组合物的制造方法与工艺
- 作为脾酪氨酸激酶抑制剂的二氮杂螺烷酮取代的噁唑衍生物的制造方法与工艺
- 包含热弹性感觉剂和TRPA1受体抑制剂的皮肤接合剃刮助剂的制造方法与工艺
- 检测EGFR耐药性突变的方法和组合物与制造工艺
- 5‑溴‑7‑氮杂吲哚啉‑2‑酮类化合物及其制备方法与制造工艺
- 3‑[(3‑{[4‑(4‑吗啉基甲基)‑1H‑吡咯‑2‑基]亚甲基}‑2‑氧代‑2,3‑二氢‑1H‑吲哚‑5‑基)甲基]‑1,3‑噻唑烷‑2,4‑二酮和EGFR TYR激酶抑制剂的组合的制造方法与工艺
- 一种抗癌药物的晶体化合物及其制备方法与制造工艺