手性(3Z,9Z)‑6,7‑环氧十八碳二烯的对映选择性合成方法与流程
本发明涉及手性(3Z,9Z)-6,7-环氧十八碳二烯的对映选择性合成方法,属于茶尺蠖防治技术领域。
背景技术:
茶尺蠖属鳞翅目(Lepidoptera)、尺蛾科(Geometriidae)、枝尺蛾亚科(Ennominae),又名拱拱虫、量寸虫、吊丝虫,为我国浙苏皖地区茶园的主要害虫。其发生代数随当年气候条件而异,一年发生5~7代,爆发时危害极重,整棵茶树叶片及嫩芽全被吃光,仅余枝干及叶脉,状如火烧,导致茶叶减产60%以上,并可导致茶树早衰、耐寒力差、冬季易受冻害等后果。茶尺蠖对茶叶产量、品质和茶树树势影响较大,对夏、秋茶为害最重;除危害茶树外,还可危害大豆、豇豆、芝麻、向日葵等。尽管农业防治和物理防治在茶尺蠖防治中仍有采用,但由于化学杀虫剂防治具有效率高、速度快、地域限制小、使用简便、防治面广等优点,因此目前对于茶尺蠖防治主要以化学防治为主。多年来,大量使用农药毒杀害虫同时导致天敌受到伤害、害虫的抗药性增强,造成了环境污染和茶叶中的农药残留,这些也直接影响了茶叶的出口贸易和人体健康。因此环境友好的生物农药逐渐成为今后害虫防治的发展方向,其中昆虫性信息素防治技术具有广泛的应用前景。
南开大学刘天麟等成功合成了七种候选化合物,室内生测实验结果表明(3Z,6Z,9Z)-十八碳三烯及(3Z,9Z)-6,7-环氧-十八碳二烯具有较强的茶尺蠖诱引活性(刘天麟,李正名等.茶尺蠖性信息素几种活性成分的合成.南开大学学报:自然科学版.1994,1:82-86)。2010年,安徽农业大学宛晓春课题组以溶剂提取法于茶尺蠖雌蛾性腺提取物中成功分离鉴定了上述两种化合物并确定了茶尺蠖性信息素的组成为(3Z,9Z)-6,7-环氧-十八碳二烯(主要组分)以及(3Z,6Z,9Z)-十八碳三烯(次要组分)(杨云秋,宛晓春等.茶尺蠖性行为习性初报.中国农学通报.2008,24:339-342.)。然而三十年来仅有两个课题组完成了茶尺蠖性信息素左旋及右旋体的对映选择性合成,合成方法上仍有一定的局限性;天然茶尺蠖性信息素的构型仍未确定,两种对映体的生物活性差异也未被研究。因此合成光学活性的茶尺蠖性信息素对于实现绿色防治、提高中国茶业竞争力具有重要意义。
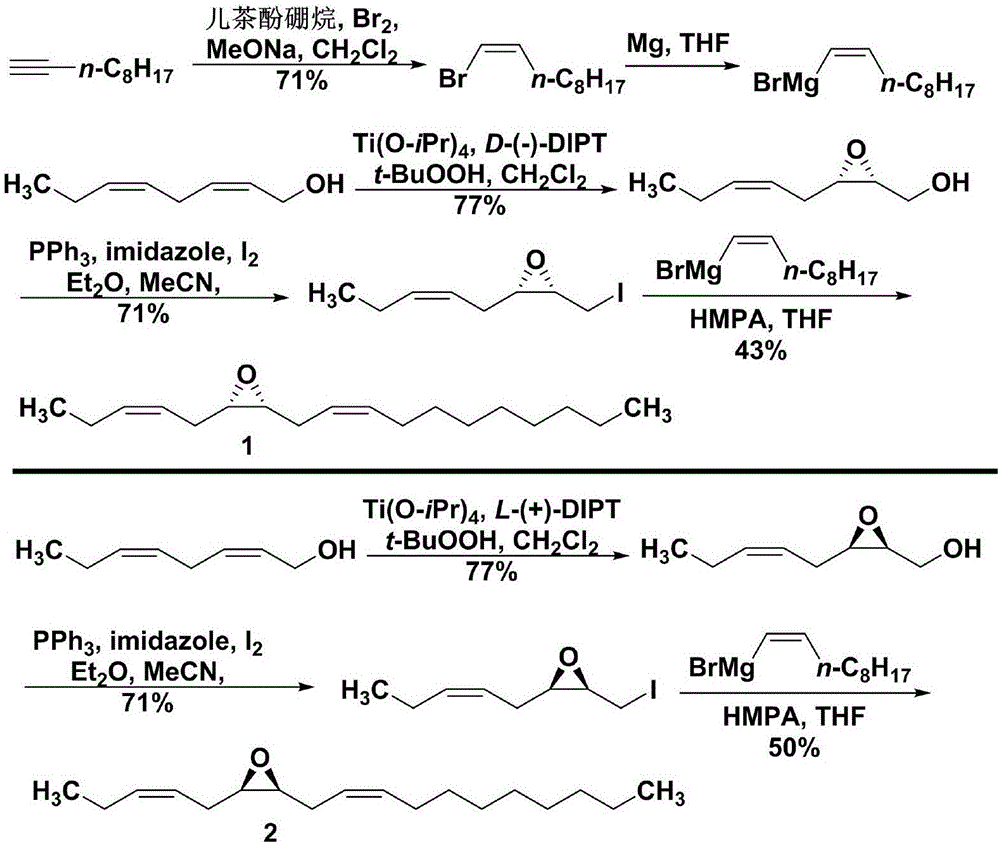
如图1所示,(3Z,9Z)-6,7-环氧十八碳二烯的光学异构体化合物1(左旋体)和2(右旋体)。
已报道的不对称合成方法如下:1986年,Millar小组(Millar,J.G.et al.Synthesis of Chiral Bis-Homoallylic Epoxides.A New Class of Lepidopteran Sex Attractants.J.Org.Chem.1986,51:4726-4728)在合成月蛾性信息素对映体系列衍生物时,从中间体(2Z,5Z)-辛二烯-1-醇出发,合成了光学活性的化合物1和2,总产率分别达到21%和26%。其合成路线如图2所示。该方法的主要缺点是顺式烯基格氏试剂难以制备,常常会有顺反异构发生;最后一步偶联反应收率低、选择性差、纯化困难,不利于大规模合成(化合物简写:Br2是溴单质,MeONa是甲醇钠,CH2Cl2是二氯甲烷,Mg是金属镁,THF是四氢呋喃,Ti(i-PrO)4是四异丙氧基钛,L-(+)-DIPT是L-酒石酸二异丙基酯,t-BuO2H是过氧叔丁醇,PPh3是三苯基膦,imidazole是咪唑,I2是碘单质,Et2O是乙醚,CH3CN是乙腈,HMPA是六甲基磷酰胺,D-(-)-DIPT是D-酒石酸二异丙基酯。)。
最近郑剑锋和黄培强等人(郑剑锋,梁攀,黄培强.一种茶尺蠖性信息素的立体选择性合成.中国发明专利.CN102911136B.)以顺-2-丁烯-1,4-二醇为起始原料,以六步反应、17%总收率获得了对映异构体1和2。其合成路线如图3所示。该方法的缺点是所用中间体1,2-二溴丁烯难以制备且极不稳定不易存储,合成中间体2,3-环氧-5-炔-十四碳醇三氟甲烷磺酸酯性质活泼,极易发生消除等反应,不利于后续反应的进行,且反应常需在低温下(零下60℃以下)进行(化合物简写:SOCl2是氯化亚砜,THF是四氢呋喃,CuI是碘化亚铜,NaI是碘化钠,K2CO3是碳酸钾,DMF是N,N-二甲基甲酰胺,1-Decyne是1-癸炔,silica gel是硅胶,CaH2是氢化钙,CH2Cl2是二氯甲烷,Tf2O是三氟甲烷磺酸酐,NEt3是三乙胺,n-BuLi是正丁基锂,n-hexane是正己烷)。
技术实现要素:
为了解决上述问题,本发明提供了一种高效、高选择性且操作简单的关于茶尺蠖性信息素左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]或者右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]的合成方法。
所述左旋体或者右旋体的合成方法,是以式(A)、式(B)、式(C)、式(D)中的任意一种或者多种化合物为起始原料或者中间体来合成左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]或者右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]。
式(A)中,2位和3位具有手性,式(A)化合物为(2R,3S)构型或(2S,3R)构型或其混合物;R1为对甲苯磺酰基,甲磺酰基,苯磺酰基,对硝基苯磺酰基等易于作为羟基离去基团的保护基,优选的R1为对甲苯磺酰基。
式(B)中,2位和3位具有手性,式(B)化合物为(2R,3R)构型或(2S,3S)构型或其混合物;R1为对甲苯磺酰基,甲磺酰基,苯磺酰基,对硝基苯磺酰基等易于作为羟基离去基团的保护基,优选的R1为对甲苯磺酰基。X为Cl,Br,I等卤素;优选的,X为Cl。
式(C)中,2位和3位具有手性,式(C)化合物为(2R,3R)构型或(2S,3S)构型或其混合物;X为Cl,Br,I等卤素;优选的,X为Cl。
式(D)中,2位和3位具有手性,式(D)化合物为(2R,3R)构型或(2S,3S)构型或其混合物;X为Cl,Br,I等卤素;优选的,X为Cl。
在一种实施方式中,所述左旋体或者右旋体的合成路线,包括如下步骤(a)至步骤(e)中的任意一个或者多个步骤:
步骤(a):
步骤(b):
步骤(c):
步骤(d):
步骤(e):式(D)化合物经关环反应、催化加氢反应得到左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]或者右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]。
在一种实施方式中,所述步骤(a)是先发生环氧化反应再与磺酸酐或磺酰氯反应得到式(A)化合物。
在一种实施方式中,所述步骤(a)是在-40~20℃、卤代烃溶剂中,在催化剂、氧化剂和添加物等作用下发生环氧化反应;然后在-20~20℃、卤代烃或醚类溶剂中,在有机碱作用下,与磺酸酐或磺酰氯反应。
在一种实施方式中,所述步骤(b)是在-40~0℃下、在卤代烃或醚类溶剂中,在路易斯酸和亲核试剂作用下发生环氧开环反应。
在一种实施方式中,所述步骤(c)是在0~30℃下,在醇类极性质子溶剂中,在无机碱作用下发生关环反应。
在一种实施方式中,所述步骤(d)是在-40~0℃下,在醚类溶剂中,在碱、路易斯酸和络合剂作用下与1-癸炔发生环氧开环反应。
在一种实施方式中,所述步骤(e)是在0~30℃下,在醇类极性质子溶剂中,在无机碱作用下发生关环反应;然后在0~30℃下,在被氢气饱和的低沸点烷烃溶剂、醚类或卤代烃溶剂中,在林德拉催化剂作用下发生催化加氢反应。
在一种实施方式中,所述左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]的合成路线如下:
所述右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]的合成路线如下:
在以下的陈述中,特定的合成产物中间体是根据结构式中的编号,用阿拉伯数字表示的,R或S表示化合物的绝对构型。
在一种实施方式中,本发明的起始原料(2Z,5Z)-辛二烯-1-醇(3)可参考文献方法(J.Org.Chem.1986,51:4726-4728)快速获得。
在一种实施方式中,本发明的左旋体1的合成方法包括以下步骤1)~7):
1)在-40~20℃下,化合物3在卤代烃溶剂中,在催化剂、氧化剂和添加物等作用下发生环氧化反应,再经过淬灭、萃取、干燥、浓缩后得到化合物4。
2)在-20~20℃下,化合物4在卤代烃或醚类溶剂中,在有机碱作用下,与磺酸酐或磺酰氯反应,再经过萃取、干燥、浓缩、纯化后得到化合物5。
3)在-40~0℃下,化合物5在卤代烃或醚类溶剂中,在路易斯酸和亲核试剂作用下发生环氧开环反应,再经过淬灭、萃取、干燥、浓缩后得到化合物6。
4)在0~30℃下,化合物6在醇类极性质子溶剂中,在无机碱作用下发生关环反应,再经过萃取、干燥、浓缩后得到化合物7。
5)在-40~0℃下,化合物7在醚类溶剂中,在碱、路易斯酸和络合剂作用下与1-癸炔发生环氧开环反应,再经过淬灭、萃取、干燥、浓缩、纯化后得到化合物8。
6)在0~30℃下,化合物8在醇类极性质子溶剂中,在无机碱作用下发生关环反应,再经过萃取、干燥、浓缩后得到化合物9。
7)在0~30℃下,化合物9在被氢气饱和的低沸点烷烃溶剂、醚类或卤代烃溶剂中,在林德拉催化剂作用下发生催化加氢反应,再经过过滤、浓缩、纯化后得到化合物1。
在一种实施方式中,本发明的右旋体2的合成方法包括以下步骤8)~14):
8)在-40~20℃下,化合物3在卤代烃溶剂中,在催化剂、氧化剂和添加物等作用下发生环氧化反应,再经过淬灭、萃取、干燥、浓缩后得到化合物10。
9)在-20~20℃下,化合物10在卤代烃或醚类溶剂中,在有机碱作用下,与磺酸酐或磺酰氯反应,再经过萃取、干燥、浓缩、纯化后得到化合物11。
10)在-40~0℃下,化合物11在卤代烃或醚类溶剂中,在路易斯酸和亲核试剂作用下发生环氧开环反应,再经过淬灭、萃取、干燥、浓缩后得到化合物12。
11)在0~30℃下,化合物12在醇类极性质子溶剂中,在无机碱作用下发生关环反应,再经过萃取、干燥、浓缩后得到化合物13。
12)在-40~0℃下,化合物13在醚类溶剂中,在碱、路易斯酸和络合剂作用下与1-癸炔发生环氧开环反应,再经过淬灭、萃取、干燥、浓缩、纯化后得到化合物14。
13)在0~30℃下,化合物14在醇类极性质子溶剂中,在无机碱作用下发生关环反应,再经过萃取、干燥、浓缩后得到化合物15。
14)在0~30℃下,化合物15在被氢气饱和的低沸点烷烃溶剂、醚类或卤代烃溶剂中,在林德拉催化剂作用下发生催化加氢反应,再经过过滤、浓缩、纯化后得到化合物2。
在一种实施方式中,所述卤代烃溶剂(步骤1)、步骤2)、步骤3)、步骤7)、步骤8)、步骤9)、步骤10)或者步骤14)中),可选自C1~C4的卤代烃,特别是二氯甲烷或三氯甲烷等。
在一种实施方式中,在步骤1)(或者步骤(a))中,所述催化剂为钛催化剂,特别是钛酸四异丙酯和D-酒石酸酯形成的金属配合物催化剂。
在一种实施方式中,在步骤1)(或者步骤(a))中,所述D-酒石酸酯为D-酒石酸二异丙酯和/或D-酒石酸二乙酯。
在一种实施方式中,在步骤1)(或者步骤(a))中,所述化合物3:钛酸四异丙酯:D-酒石酸酯:过氧叔丁醇的摩尔比可为1:1:1.1:2。
在一种实施方式中,在步骤1)((或者步骤(a)、步骤8))中,所述氧化剂可为无水过氧叔丁醇的壬烷溶液。
在一种实施方式中,在步骤1)(或者步骤(a)、步骤8))中,所述添加物的主要作用是吸水。
在一种实施方式中,在步骤1)(或者步骤(a)、步骤8))中,所述添加物为分子筛、分子筛、分子筛、100~200目或300~400目的硅胶和氢化钙等。
在一种实施方式中,所述醚类溶剂(步骤(a)、步骤2)、步骤3)、步骤5)、步骤7)、步骤9)、步骤10)、步骤12)、或者步骤14)中)选自C2~C4的脂肪醚或脂环醚等,特别是乙醚或四氢呋喃等。
在一种实施方式中,在步骤2)(或者步骤(a)、步骤9))中,所述有机碱可选用叔胺等,特别是咪唑、吡啶、三乙胺、二异丙基乙基胺、二氮杂二环(DBU)或三乙烯二胺(DABCO)等。
在一种实施方式中,在步骤2)(或者步骤(a)、步骤9))中,所述磺酸酐可选用三氟甲烷磺酸酐等,所述磺酰氯可选用甲烷磺酰氯、对甲苯磺酰氯、苯磺酰氯、对硝基苯磺酰氯等。
在一种实施方式中,在步骤2)(或者步骤(a)、步骤9))中,所述化合物4(或者化合物10):有机碱:磺酸酐或磺酰氯的摩尔比可为1:2.0:1.5。
在一种实施方式中,在步骤3)(或者步骤(b)、步骤10))中,所述路易斯酸可选用烷基氯化铝、烷基氯化镁等,特别是二甲基氯化铝、二乙基氯化铝、二正丁基氯化铝、二异丁基氯化铝等。
在一种实施方式中,在步骤3)(或者步骤(b)、步骤10))中,所述化合物5(或者式(A)化合物、化合物11):路易斯酸的摩尔比可为1:2.0。
在一种实施方式中,所述醇类极性质子溶剂(步骤4)、步骤(c)、步骤6)、步骤11)或者步骤13)中)可选自C1~C4的脂肪醇,特别是甲醇或乙醇等;
在一种实施方式中,所述步骤4)、步骤(c)、步骤6)、步骤11)或者步骤13)中的无机碱可为碳酸钠、碳酸钾或碳酸铯等。
在一种实施方式中,在步骤4)(或者步骤c)、步骤11))中,所述化合物6(或者式(B)化合物、化合物12):无机碱的摩尔比可为1:2.0。
在一种实施方式中,在步骤5)(或者步骤(d)、步骤12))中,所述路易斯酸可为三氟化硼乙醚。
在一种实施方式中,在步骤5)(或者步骤(d)、步骤12))中,所述络合剂可为六甲基磷酰三胺。
在一种实施方式中,在步骤5)(或者步骤(d)、步骤12))中,碱可为正丁基锂、仲丁基锂或叔丁基锂。
在一种实施方式中,在步骤5)(或者步骤(d)、步骤12))中,所述化合物7(或者式(C)化合物、化合物13):碱:路易斯酸:络合剂:1-癸炔的摩尔比可为1:3.0:3.0:3.0:2.0。
在一种实施方式中,在步骤6)(或者步骤(e)、步骤13))中,所述化合物8(或者式(D)化合物、化合物14):无机碱的摩尔比可为1:3.0。
在一种实施方式中,在步骤7)(或者步骤(e)、步骤14))中,所述低沸点烷烃溶剂可选自C5~C6的脂肪族饱和烷烃等,特别是正戊烷、环己烷或正己烷等。
在一种实施方式中,在步骤7)(或者步骤(e)、步骤14))中,所述林德拉催化剂可选自金属铅中毒或喹啉中毒的金属钯等,催化剂载体是碳酸钙或硫酸钡等。
在一种实施方式中,在步骤7)(或者步骤(e)、步骤14))中,所述化合物9(或者化合物15):催化剂的质量分数比可为10:1。
在一种实施方式中,在步骤8)(或者步骤(a))中,所述催化剂为钛催化剂,特别是钛酸四异丙酯和L-酒石酸酯形成的金属配合物催化剂。
在一种实施方式中,在步骤8)(或者步骤(a)),所述化合物3:钛酸四异丙酯:L-酒石酸酯:过氧叔丁醇的摩尔比可为1:1:1.1:2。
在一种实施方式中,所述L-酒石酸酯为L-酒石酸二异丙酯和/或L-酒石酸二乙酯。
本发明还提供所述方法在茶尺蠖防治方面的应用。
本发明通过对光学活性的左旋体1和右旋体2进行生物活性测试,并确定两种对映异构体之间的活性差异。
本发明还提供一种茶尺蠖触角电位试剂,所述试剂以左旋体[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]为有效成分或者主要有效成分。
在一种实施方式中,所述试剂的有效成分中左旋体[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]的质量分数为试剂的95%以上、92%以上、93%~98%、72%~85%等。
在一种实施方式中,所述试剂中还含有(3Z,6Z,9Z)十八碳三烯。
本发明的优点和效果:
(1)本发明以易得的(2Z,5Z)-辛二烯-1-醇(3)为起始原料,高效、高立体选择性地合成了茶尺蠖性信息素左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]及右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]。
(2)本发明各步骤操作简单,纯化简单,很多步骤不需纯化即可直接进行下步反应,所用试剂均为常用易得廉价试剂,路线较短,适合大规模生产。
(3)本发明经7步总收率27%合成了(-)-(3Z,9Z,6S,7R)-1,对映选择性(ee)高达99%;经7步总收率24%合成了(+)-(3Z,9Z,6R,7S)-2,对映选择性(ee)高达98%。
(4)发明通过EAG(触角电位)实验,发现茶尺蠖对左旋体1的触角电位反应明显要高于右旋体2,可制备成茶尺蠖诱捕剂。
附图说明:
图1:(3Z,9Z)-6,7-环氧十八碳二烯的光学异构体化合物;
图2:(3Z,9Z)-6,7-环氧十八碳二烯的光学异构体的一种合成路线;
图3:(3Z,9Z)-6,7-环氧十八碳二烯的光学异构体的另一种合成路线;
图4::单一化合物对茶尺蠖的触角电位反应图;其中环氧化合物与(3Z,6Z,9Z)十八碳三烯的配比为6:4;
图5:二元混合物对茶尺蠖的触角电位反应图;其中环氧化合物与(3Z,6Z,9Z)十八碳三烯的配比为6:4。
具体实施方案
下面结合具体实施例对本发明作进一步详细阐述。
实施例1
步骤1)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇(4)
在氮气氛保护下,用500mg硅胶混合200mg氢化钙,加入二氯甲烷(25mL)溶剂和钛酸四异丙酯(3.0mL,10mmol),置于-30℃下搅拌,再依次加入D-(-)-酒石酸二乙酯(2.27g,11mmol)的二氯甲烷(10mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.26g,10mmol)的二氯甲烷(5mL)溶液、过氧叔丁醇(3.7mL,20mmol,5.5M in n-Nonane),随后于-30℃反应2天。用30mL10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物4,无需纯化直接进行下一步反应。
步骤2)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(5)
将粗产品4溶于二氯甲烷(30mL)中,于0℃下加入三乙胺(2.8mL,20mmol)和对甲苯磺酰氯(2.86g,15mmol),继续搅拌并缓慢回复至室温,用30mL饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得化合物5(1.81g,两步收率61%),无色液体,[α]D20=+15.34(c=1.74in CH2Cl2);1H NMR(400MHz,CDCl3)δ7.74(d,J=8.0Hz,2H),7.29(d,J=8.0Hz,2H),5.51-5.38(m,1H),5.29-5.15(m,1H),4.14(dd,J=11.2,4.8Hz,1H),4.03(dd,J=11.2,6.5Hz,1H),3.14-3.03(m,1H),2.99-2.87(m,1H),2.38(s,3H),2.26-2.16(m,1H),2.09-1.99(m,1H),1.97-1.86(m,2H),0.88(t,J=7.5Hz,3H);13C NMR(101MHz,CDCl3)δ145.18,134.98,132.58,129.94,127.96,122.13,68.08,55.92,53.02,26.01,21.64,20.65,14.07;IR(KBr):γ3060,2960,2932,1625,1448,1250,1098,795,691cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:297.11605,found 297.11529.
步骤3)合成(2R,3R,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(6)
在氮气氛保护下,将化合物5(1.48g,5mmol)溶于无水四氢呋喃(50mL)中,在-40℃下滴加二乙基氯化铝(10mL,10mmol,1.0M in n-hexane)。继续在相同温度下反应半小时后,用10mL饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯(3×20mL)萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物6,无需纯化直接进行下一步反应。
步骤4)合成(2R,3R,5Z)-1,2-环氧-3-氯-5-辛烯(7)
将粗产品6溶于甲醇(30mL)中,在0℃下加入无水碳酸钠(1.06g,10mmol),反应一小时后,溶剂经减压浓缩后加入30mL水和20mL二氯甲烷,分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物7,无需纯化直接进行下一步反应。
步骤5)合成(6R,7R,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(8)
在氮气氛保护下,将1-癸炔(1.38g,10mmol)溶于无水四氢呋喃(30mL)中,在-40℃下滴加正丁基锂(6.3mL,15mmol,2.4M in n-hexane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液(1.9mL,15mmol)和六甲基磷酰三胺(2.6mL,15mmol),再反应十分钟后滴加粗产品7的四氢呋喃溶液(10mL)。继续在相同温度下反应半小时后,用10mL饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯(3×20mL)萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得化合物8(942mg,三步收率63%),无色液体,[α]D20=+9.44(c=1.48in CH2Cl2);1H NMR(400MHz,CDCl3)δ5.49(dd,J=17.0,7.3Hz,1H),5.32(dd,J=17.0,8.0Hz,1H),4.12(dd,J=9.4,4.5Hz,1H),3.75(d,J=5.6Hz,1H),2.67-2.34(m,4H),2.13-1.98(m,4H),1.48-1.35(m,2H),1.30-1.15(m,10H),0.91(t,J=7.5Hz,3H),0.81(t,J=6.4Hz,3H);13C NMR(101MHz,CDCl3)δ135.30,123.84,83.48,75.08,71.78,66.01,32.85,31.92,29.29,29.18,28.96,25.36,22.74,20.84,18.79,14.17,14.14;IR(KBr):γ3055,2960,2932,2205,1621,1430,1098,693cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:299.21417,found 299.21459.
步骤6)合成(6S,7R,3Z)-十八碳-3-烯-9-炔(9)
将化合物8(600mg,2.0mmol)溶于甲醇(15mL)中,在0℃下加入无水碳酸钠(636mg,6mmol)。继续反应一小时后,溶剂经减压浓缩后加入30mL水和50mL二氯甲烷,分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物9,无需纯化直接进行下一步反应。
步骤7)合成左旋-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯(1)
在室温和氢气氛下,向林德拉催化剂(Pd/BaSO4/喹啉,50mg,10wt%)中加入化合物9的二氯甲烷(20mL)溶液,反应两小时后过滤出去催化剂,滤饼用二氯甲烷洗涤,滤液经浓缩纯化后得无色液体1(375mg,两步收率71%)。[α]D20=-1.29(c=1.5in CH2Cl2);1H NMR(400MHz,CDCl3)δ5.60-5.50(m,2H),5.47-5.37(m,2H),2.97-2.91(m,2H),2.45-2.36(m,2H),2.26-2.16(m,2H),2.09-1.99(m,4H),1.37-1.26(m,12H),0.99(t,J=7.5Hz,3H),0.87(t,J=7.0Hz,3H);13C-NMR(CDCl3,101MHz)δ(ppm):134.49,133.01,123.21,122.58,57.62,31.81,29.65,29.49,29.18,27.33,22.65,14.15,14.01;IR(KBr):γ2956,2921,1734,1657,1459,1260,1015cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:265.25314,found 265.25247.
步骤8)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇(10)
在氮气氛保护下,用600mg硅胶混合300mg氢化钙,加入二氯甲烷(35mL)溶剂和钛酸四异丙酯(4.5mL,15mmol),置于-30℃下搅拌,再依次加入L-(+)-酒石酸二乙酯(3.4g,16.5mmol)的二氯甲烷(15mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.89g,15mmol)的二氯甲烷(10mL)溶液、过氧叔丁醇(5.5mL,30mmol,5.5M in n-Nonane),随后于-30℃反应2天。用30mL10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷(2×30mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物10,无需纯化直接进行下一步反应。
步骤9)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(11)
将粗产品10溶于二氯甲烷(40mL)中,于0℃下加入三乙胺(4.2mL,30mmol)和对甲苯磺酰氯(4.29g,22.5mmol),继续搅拌并缓慢回复至室温,用30mL饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷(2×30mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得化合物11(2.62g,两步收率59%),无色液体,[α]D20=-14.88(c=1.69in CH2Cl2);1H NMR(400MHz,CDCl3)δ7.74(d,J=8.0Hz,2H),7.29(d,J=8.0Hz,2H),5.51-5.38(m,1H),5.29-5.15(m,1H),4.14(dd,J=11.2,4.8Hz,1H),4.03(dd,J=11.2,6.5Hz,1H),3.14-3.03(m,1H),2.99-2.87(m,1H),2.38(s,3H),2.26-2.16(m,1H),2.09-1.99(m,1H),1.97-1.86(m,2H),0.88(t,J=7.5Hz,3H);13C NMR(101MHz,CDCl3)δ145.18,134.98,132.58,129.94,127.96,122.13,68.08,55.92,53.02,26.01,21.64,20.65,14.07;IR(KBr):γ3060,2960,2932,1625,1448,1250,1098,795,691cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:297.11605,found 297.11544.
步骤10)合成(2S,3S,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(12)
在氮气氛保护下,将化合物11(1.48g,5mmol)溶于无水四氢呋喃(50mL)中,在-40℃下滴加二乙基氯化铝(10mL,10mmol,1.0M in n-hexane)。继续在相同温度下反应半小时后,用10mL饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯(3×20mL)萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物12,无需纯化直接进行下一步反应。
步骤11)合成(2S,3S,5Z)-1,2-环氧-3-氯-5-辛烯(13)
将粗产品12溶于甲醇(30mL)中,在0℃下加入无水碳酸钠(1.06g,10mmol),反应一小时后,溶剂经减压浓缩后加入30mL水和20mL二氯甲烷,分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物13,无需纯化直接进行下一步反应。
步骤12)合成(6S,7S,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(14)
在氮气氛保护下,将1-癸炔(1.38g,10mmol)溶于无水四氢呋喃(30mL)中,在-40℃下滴加正丁基锂(6.3mL,15mmol,2.4M in n-hexane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液(1.9mL,15mmol)和六甲基磷酰三胺(2.6mL,15mmol),再反应十分钟后滴加粗产品13的四氢呋喃溶液(10mL)。继续在相同温度下反应半小时后,用10mL饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯(3×20mL)萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得化合物14(897mg,三步收率60%),无色液体,[α]D20=-9.12(c=1.40in CH2Cl2);1H NMR(400MHz,CDCl3)δ5.49(dd,J=17.0,7.3Hz,1H),5.32(dd,J=17.0,8.0Hz,1H),4.12(dd,J=9.4,4.5Hz,1H),3.75(d,J=5.6Hz,1H),2.67-2.34(m,4H),2.13-1.98(m,4H),1.48-1.35(m,2H),1.30-1.15(m,10H),0.91(t,J=7.5Hz,3H),0.81(t,J=6.4Hz,3H);13C NMR(101MHz,CDCl3)δ135.30,123.84,83.48,75.08,71.78,66.01,32.85,31.92,29.29,29.18,28.96,25.36,22.74,20.84,18.79,14.17,14.14;IR(KBr):γ3055,2960,2932,2205,1621,1430,1098,693cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:299.21417,found 299.21463.
步骤13)合成(6R,7S,3Z)-十八碳-3-烯-9-炔(15)
将化合物14(600mg,2.0mmol)溶于甲醇(15mL)中,在0℃下加入无水碳酸钠(636mg,6mmol)。继续反应一小时后,溶剂经减压浓缩后加入30mL水和50mL二氯甲烷,分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相再用饱和食盐水(3×30mL)洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物15,无需纯化直接进行下一步反应。
步骤14)合成右旋-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯(2)
在室温和氢气氛下,向林德拉催化剂(Pd/BaSO4/喹啉,50mg,10wt%)中加入化合物15的二氯甲烷(20mL)溶液,反应两小时后过滤出去催化剂,滤饼用二氯甲烷洗涤,滤液经浓缩纯化后得无色液体2(364mg,两步收率69%)。[α]D20=+1.62(c=1.1in CH2Cl2);1H NMR(400MHz,CDCl3)δ5.60-5.50(m,2H),5.47-5.37(m,2H),2.97-2.91(m,2H),2.45-2.36(m,2H),2.26-2.16(m,2H),2.09-1.99(m,4H),1.37-1.26(m,12H),0.99(t,J=7.5Hz,3H),0.87(t,J=7.0Hz,3H);13C-NMR(CDCl3,101MHz)δ(ppm):134.49,133.01,123.21,122.58,57.62,31.81,29.65,29.49,29.18,27.33,22.65,14.15,14.01;IR(KBr):γ2956,2921,1734,1657,1459,1260,1015cm-1;HRMS(ESI)calculated for C18H20NO[M+H]+:265.25314,found 265.25247.
实施例2
步骤1)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇(4)
在氮气氛保护下,向500mg分子筛加入二氯甲烷(25mL)溶剂和钛酸四异丙酯(3.0mL,10mmol),置于-30℃下搅拌,再依次加入D-(-)-酒石酸二乙酯(2.27g,11mmol)的二氯甲烷(10mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.26g,10mmol)的二氯甲烷(5mL)溶液、过氧叔丁醇(3.7mL,20mmol,5.5M in n-Nonane),随后于-30℃反应2天。用10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物4,无需纯化直接进行下一步反应。
步骤2)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(5)
将粗产品4溶于吡啶(30mL)中,于0℃下加入对甲苯磺酰氯,继续搅拌并缓慢回复至室温,用饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷(2×20mL)萃取,合并的有机相依次用1M盐酸、饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得无色液体化合物5(两步收率56%)。
步骤3)合成(2R,3R,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(6)
在氮气氛保护下,将化合物5溶于无水乙醚中,在-40℃下滴加二甲基氯化铝(0.9M in heptane)。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物6,无需纯化直接进行下一步反应。
步骤4)合成(2R,3R,5Z)-1,2-环氧-3-氯-5-辛烯(7)
将粗产品6溶于甲醇中,在0℃下加入无水碳酸钾,反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物7,无需纯化直接进行下一步反应。
步骤5)合成(6R,7R,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(8)
在氮气氛保护下,将1-癸炔溶于无水四氢呋喃中,在-40℃下滴加叔丁基锂(1.6M in pentane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液和六甲基磷酰三胺,再反应十分钟后滴加粗产品7的四氢呋喃溶液。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得无色液体化合物8(三步收率55%)。
步骤6)合成(6S,7R,3Z)-十八碳-3-烯-9-炔(9)
将化合物8溶于甲醇中,在0℃下加入无水碳酸钾。继续反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物9,无需纯化直接进行下一步反应。
步骤7)合成左旋-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯(1)
在0℃和氢气氛下,向林德拉催化剂(Pd/CaCO3/Pd2+,10wt%)中加入化合物9的乙醚溶液,反应两小时后过滤出去催化剂,滤饼用乙醚洗涤,滤液经浓缩纯化后得无色液体1(两步收率73%)。
步骤8)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇(10)
在氮气氛保护下,向700mg分子筛加入二氯甲烷(35mL)溶剂和钛酸四异丙酯(4.5mL,15mmol),置于-30℃下搅拌,再依次加入L-(+)-酒石酸二乙酯(3.4g,16.5mmol)的二氯甲烷(15mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.89g,15mmol)的二氯甲烷(10mL)溶液、过氧叔丁醇(5.5mL,30mmol,5.5M in n-Nonane),随后于-30℃反应2天。用10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物10,无需纯化直接进行下一步反应。
步骤9)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(11)
将粗产品10溶于吡啶(30mL)中,于0℃下加入对甲苯磺酰氯,继续搅拌并缓慢回复至室温,用饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷萃取,合并的有机相依次用1M盐酸、饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得化合物11(两步收率57%)。
步骤10)合成(2S,3S,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(12)
在氮气氛保护下,将化合物11溶于无水乙醚中,在-40℃下滴加二甲基氯化铝(0.9M in heptane)。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物12,无需纯化直接进行下一步反应。
步骤11)合成(2S,3S,5Z)-1,2-环氧-3-氯-5-辛烯(13)
将粗产品12溶于甲醇中,在0℃下加入无水碳酸钾,反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物13,无需纯化直接进行下一步反应。
步骤12)合成(6S,7S,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(14)
在氮气氛保护下,将1-癸炔溶于无水四氢呋喃中,在-40℃下滴加叔丁基锂(1.6M in pentane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液和六甲基磷酰三胺,再反应十分钟后滴加粗产品13的四氢呋喃溶液。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得化合物14(三步收率62%)。
步骤13)合成(6R,7S,3Z)-十八碳-3-烯-9-炔(15)
将化合物14溶于甲醇中,在0℃下加入无水碳酸钾。继续反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物15,无需纯化直接进行下一步反应。
步骤14)合成右旋-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯(2)
在0℃和氢气氛下,向林德拉催化剂(Pd/CaCO3/Pd2+,10wt%)中加入化合物15的乙醚溶液,反应两小时后过滤出去催化剂,滤饼用乙醚洗涤,滤液经浓缩纯化后得无色液体2(两步收率70%)。
实施例3
步骤1)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇(4)
在氮气氛保护下,向500mg分子筛加入二氯甲烷(25mL)溶剂和钛酸四异丙酯(3.0mL,10mmol),置于-30℃下搅拌,再依次加入D-(-)-酒石酸二乙酯(2.27g,11mmol)的二氯甲烷(10mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.26g,10mmol)的二氯甲烷(5mL)溶液、过氧叔丁醇(3.7mL,20mmol,5.5M in n-Nonane),随后于-30℃反应2天。用10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物4,无需纯化直接进行下一步反应。
步骤2)合成(2R,3S,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(5)
将粗产品4溶于无水四氢呋喃中,于0℃下加入咪唑和对甲苯磺酰氯,继续搅拌并缓慢回复至室温,用饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷萃取,合并的有机相依次用1M盐酸、饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得无色液体化合物5(两步收率64%)。
步骤3)合成(2R,3R,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(6)
在氮气氛保护下,将化合物5溶于二氯甲烷中,在-40℃下滴加二异丁基氯化铝(0.8M in heptane)。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用二氯甲烷萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物6,无需纯化直接进行下一步反应。
步骤4)合成(2R,3R,5Z)-1,2-环氧-3-氯-5-辛烯(7)
将粗产品6溶于无水乙醇中,在0℃下加入无水碳酸钾,反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物7,无需纯化直接进行下一步反应。
步骤5)合成(6R,7R,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(8)
在氮气氛保护下,将1-癸炔溶于无水乙醚中,在-40℃下滴加正丁基锂(2.4M in n-hexane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液和六甲基磷酰三胺,再反应十分钟后滴加粗产品7的四氢呋喃溶液。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得无色液体化合物8(三步收率57%)。
步骤6)合成(6S,7R,3Z)-十八碳-3-烯-9-炔(9)
将化合物8溶于无水乙醇中,在0℃下加入无水碳酸钾。继续反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物9,无需纯化直接进行下一步反应。
步骤7)合成左旋-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯(1)
在0℃和氢气氛下,向林德拉催化剂(Pd/BaSO4/喹啉,10wt%)中加入化合物9的石油醚溶液,反应两小时后过滤出去催化剂,滤饼用乙醚洗涤,滤液经浓缩纯化后得无色液体1(两步收率72%)。
步骤8)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇(10)
在氮气氛保护下,向700mg分子筛加入二氯甲烷(35mL)溶剂和钛酸四异丙酯(4.5mL,15mmol),置于-30℃下搅拌,再依次加入L-(+)-酒石酸二乙酯(3.4g,16.5mmol)的二氯甲烷(15mL)溶液、(2Z,5Z)-辛二烯-1-醇(3)(1.89g,15mmol)的二氯甲烷(10mL)溶液、过氧叔丁醇(5.5mL,30mmol,5.5M in n-Nonane),随后于-30℃反应2天。用10%的柠檬酸水溶液淬灭反应后,升温,静置分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩后得粗产品化合物10,无需纯化直接进行下一步反应。
步骤9)合成(2S,3R,5Z)-2,3-环氧-5-辛烯-1-醇对甲苯磺酸酯(11)
将粗产品10溶于四氢呋喃中,于0℃下加入咪唑和对甲苯磺酰氯,继续搅拌并缓慢回复至室温,用饱和氯化铵溶液淬灭反应后,静置分液,水相用二氯甲烷萃取,合并的有机相依次用1M盐酸、饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩纯化后得化合物11(两步收率58%)。
步骤10)合成(2S,3S,5Z)-2-羟基-3-氯-5-辛烯-1-醇对甲苯磺酸酯(12)
在氮气氛保护下,将化合物11溶于无二氯甲烷中,在-40℃下滴加二异丁基氯化铝(0.8M in heptane)。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用二氯甲烷萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩获得化合物12,无需纯化直接进行下一步反应。
步骤11)合成(2S,3S,5Z)-1,2-环氧-3-氯-5-辛烯(13)
将粗产品12溶于无水乙醇中,在0℃下加入无水碳酸钾,反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物13,无需纯化直接进行下一步反应。
步骤12)合成(6S,7S,3Z)-6-氯-十八碳-3-烯-9-炔-7-醇(14)
在氮气氛保护下,将1-癸炔溶于无水乙醚中,在-40℃下滴加正丁基锂(2.4M in n-hexane)。在相同温度下反应十分钟后加入三氟化硼乙醚溶液和六甲基磷酰三胺,再反应十分钟后滴加粗产品13的四氢呋喃溶液。继续在相同温度下反应半小时后,用饱和氯化铵溶液淬灭反应,分液,水相用乙酸乙酯萃取。合并的有机相用无水硫酸钠干燥,过滤浓缩纯化后得化合物14(三步收率62%)。
步骤13)合成(6R,7S,3Z)-十八碳-3-烯-9-炔(15)
将化合物14溶于无水乙醇中,在0℃下加入无水碳酸钾。继续反应一小时后,溶剂经减压浓缩后加入水和二氯甲烷,分液,水相用二氯甲烷萃取,合并的有机相再用饱和食盐水洗涤,经无水硫酸钠干燥,过滤浓缩得粗产品化合物15,无需纯化直接进行下一步反应。
步骤14)合成右旋-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯(2)
在0℃和氢气氛下,向林德拉催化剂(Pd/BaSO4/喹啉,10wt%)中加入化合物15的石油醚溶液,反应两小时后过滤出去催化剂,滤饼用乙醚洗涤,滤液经浓缩纯化后得无色液体2(两步收率67%)。
实施例4:
本发明通过EAG(触角电位)实验对合成的茶尺蠖性信息素左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]、右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]以及已知的茶尺蠖性信息素外消旋体等化合物进行了生物活性测试,结果见图4-图5所示。结果表明,无论是纯的左旋体、右旋体和外消旋体,还是这几种异构体与(3Z,6Z,9Z)-十八碳三烯(triene)的配比混合物,茶尺蠖对左旋体1的触角电位反应明显要高于右旋体2。
本发明还通过风洞实验对合成的茶尺蠖性信息素左旋体1[(-)-(3Z,9Z,6S,7R)-6,7-环氧十八碳二烯]、右旋体2[(+)-(3Z,9Z,6R,7S)-6,7-环氧十八碳二烯]以及已知的茶尺蠖性信息素外消旋体等化合物进行了生物活性测试,结果见表1。结果同样表明左旋体1诱捕茶尺蠖的活性明显高于右旋体2。
表1不同诱捕剂对茶尺蠖的风洞实验
由于天然茶尺蠖性信息素难以分离鉴定其绝对构型,通过以上生物活性实验可以初步判断本发明合成的左旋体1具有更好的诱捕茶尺蠖的能力,而右旋体2对于茶尺蠖基本没有诱捕活性。
以上所述仅为本发明的优选实例而已,并不用于限制本发明,尽管参照前述实例对本发明进行了详细说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!