一种后处理制备抗污染正渗透聚酰胺复合膜的方法与流程
本发明涉及一种后处理改性提高正渗透聚酰胺复合膜抗污染的方法。
背景技术:
水资源短缺问题已经成为全球性的挑战。膜技术具有高效率、低能耗、操作管理方便等诸多优势,在应对该挑战的过程中发挥着重要作用。作为一种新兴的渗透压驱动膜处理技术,正渗透(Forward Osmosis,FO)技术以其在处理一些特殊原水时独特优势,被认为是常规膜工艺的有效补充。正渗透技术利用汲取液和原料液之间的渗透压差作为驱动力,水通过选择性膜而将污染物等截留。相对于传统压力驱动膜工艺,正渗透技术具有污染率低,回收率高等优势,在处理具有高盐度,高污染的原料液如海水淡化,污废水回用等方面具有广泛的应用前景。
膜材料是FO技术的核心,而高性能聚酰胺复合薄膜(TFC)的出现,对正渗透的发展具有里程碑式的意义。正渗透聚酰胺复合膜由支撑层和活性层组成,所述的活性层为聚酰胺层,但是聚酰胺由于其表面本身的物化性质,如较为疏水,粗糙度较高,表面富含羧基官能团等,很容易被污染。而膜污染会导致出水量降低,水质变差,能耗增高等一系列缺点,因此,提高正渗透聚酰胺复合膜的抗污染效能,对FO技术的发展进步至关重要。
在中国已公开专利“一种后处理改性制备高选择性正渗透聚酰胺复合膜的方法”(申请号:201410815728.X)中给出一种高选择性正渗透聚酰胺复合膜的改性方法,首先制备正渗透聚酰胺复合膜,同时通过浸泡胺类溶液的后处理方式改变聚酰胺表面的电荷密度(降低羟基密度,增加胺类有机官能团),从而提高了正渗透膜对水中离子的选择性,降低正渗透的溶质返混现象。改性后的高选择性正渗透聚酰胺复合膜虽然提高了正渗透膜对水中离子的选择性,降低正渗透的溶质返混现象,但是改性后的高选择性正渗透聚酰胺复合膜在水处理过程中仍然存在易被有机污染,从而导致膜出水通量降低,出水水质变差,缩短膜生命周期,进而导致降低正渗透工艺处理效能,增加操作成本的问题。
技术实现要素:
本发明的目的是要解决现有正渗透聚酰胺膜在水处理过程中易被有机污染,导致降低正渗透工艺处理效能的问题,而提供一种后处理制备抗污染正渗透聚酰胺复合膜的方法。
一种后处理制备抗污染正渗透聚酰胺复合膜的方法,具体是按以下步骤完成的:
一、耦合:将盐酸多巴胺溶解于二甲基甲酰胺中,然后在氮气保护下加入2-溴异丁酰溴和三乙胺,并在氮气保护下搅拌反应2h~6h,得到耦合溶液;
二、接枝引发剂:向耦合溶液中加入三羟甲基氨基甲烷缓冲液,得到混合溶液,然后将正渗透聚酰胺复合膜表面的活性层浸没在混合溶液中,且保证正渗透聚酰胺复合膜表面的支撑层不与混合溶液接触,浸没时间为10min~90min,得到接枝引发剂正渗透聚酰胺复合膜;
三、接枝两性离子聚合物:将两性离子单体溶于异丙醇水溶液中,然后在氮气保护下加入氯化铜催化复合物,得到含两性离子单体溶液,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入抗坏血酸溶液,并在氮气保护下聚合反应0.5h~3h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜。
本发明的反应过程:在步骤一中,在氮气保护下,首先将原子转移自由基聚合反应将要使用的引发剂耦合接枝到多巴胺上;在步骤二中,在空气和缓冲溶液的环境下使多巴胺接触正渗透聚酰胺复合膜表面,通过多巴胺自生形成聚合多巴胺的聚合反应,使引发剂接枝在正渗透聚酰胺复合膜表面;在步骤三中,主要依靠原子转移自由基聚合反应,使两性离子单体从引发剂的位置在正渗透聚酰胺复合膜上生长,通过改变反应时间,可调控接枝的两性离子聚合物改性层。
本发明的机理:由于正渗透聚酰胺复合膜的聚酰胺活性层由界面聚合生成,其中没有反应的酰氯基发生水解反应形成的羧基为正渗透聚酰胺复合膜表面主导性官能团,导致正渗透聚酰胺复合膜表面较为疏水,粗糙度高,在水处理过程中易于被污染,同时,羧基在有钙离子存在的情况下通过架桥作用会加剧膜污染,而本发明通过在正渗透聚酰胺复合膜表面接枝可调控的超亲水的两性离子聚合物改性层,可有效提高正渗透聚酰胺复合膜表面亲水性,降低粗糙度,从而降低膜污染,同时,两性离子聚合物改性层可大大降低正渗透聚酰胺复合膜表面可用的羧基官能团浓度,降低正渗透聚酰胺复合膜表面钙离子架桥导致的膜污染。
本发明的优点:一、本发明采用原子转移自由基聚合反应ATRP,其发展较为成熟,所用化学试剂常见,同时,选用的商品化的两性离子,价格相对便宜,同时,采用的是ARGET-ATRP(电子转移活化再生催化剂原子转移自由基聚合),其特点是对氧气有一定的容忍度,需要的催化剂量少,便于以后大规模工业化开发。二、本发明得到的抗污染正渗透聚酰胺复合膜在大幅度提高抗污染性能的同时,可保持其过水性能和对盐的选择性。三、由于ATRP高度的可控性,可通过调节聚合物的生长时间等反应参数,对膜的传质和抗污染表面性能进行进一步调控和优化。四、由于本发明首先借助多巴胺将引发剂接枝在TFC膜上,而多巴胺能在溶解氧的作用下发生氧化-交联反应,形成强力附着于固体材料表面的聚合多巴胺,即多巴胺除了TFC膜的聚酰胺,也可附着在各种不同的表面,因此本发明中的方法不仅适用于TFC膜,也同时适用于其他表面的抗污染改性。
本发明属于一种后处理改性方法,主要用于正渗透聚酰胺复合膜的改性处理。
附图说明
图1是实施例1操作流程示意图;
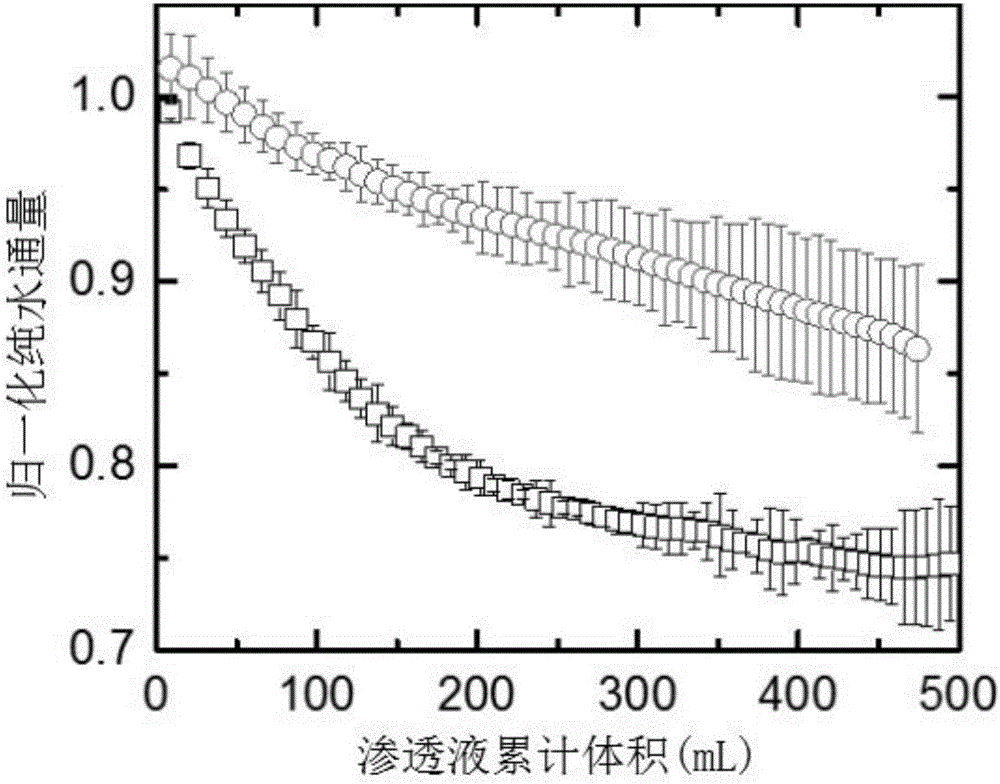
图2是实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜污染过程中纯水通量衰减示意图。
图3是水渗透系数-结盐渗透系数-构特性参数柱形图,图中A表示正渗透聚酰胺复合膜的水渗透系数柱形图,图中B表示正渗透聚酰胺复合膜的盐渗透系数柱形图,图中S表示正渗透聚酰胺复合膜的结构特性参数柱形图,图中a表示实施例1得到的抗污染正渗透聚酰胺复合膜的水渗透系数柱形图,图中b表示实施例1得到的抗污染正渗透聚酰胺复合膜的盐渗透系数柱形图,图中s表示实施例1得到的抗污染正渗透聚酰胺复合膜的结构特性参数柱形图。
具体实施方式
具体实施方式一:本实施方式是一种后处理制备抗污染正渗透聚酰胺复合膜的方法,具体是按以下步骤完成的:
一、耦合:将盐酸多巴胺溶解于二甲基甲酰胺中,然后在氮气保护下加入2-溴异丁酰溴和三乙胺,并在氮气保护下搅拌反应2h~6h,得到耦合溶液;
二、接枝引发剂:向耦合溶液中加入三羟甲基氨基甲烷缓冲液,得到混合溶液,然后将正渗透聚酰胺复合膜表面的活性层浸没在混合溶液中,且保证正渗透聚酰胺复合膜表面的支撑层不与混合溶液接触,浸没时间为10min~90min,得到接枝引发剂正渗透聚酰胺复合膜;
三、接枝两性离子聚合物:将两性离子单体溶于异丙醇水溶液中,然后在氮气保护下加入氯化铜催化复合物,得到含两性离子单体溶液,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入抗坏血酸溶液,并在氮气保护下聚合反应0.5h~3h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜。
本实施方式步骤二中所述的正渗透聚酰胺复合膜为高选择性正渗透聚酰胺复合膜,具体是按中国已公开专利“一种高选择性正渗透聚酰胺复合膜的原位制备方法”(申请号:201410815730.7)提供方法制备的。
本实施方式主要目的是提高正渗透聚酰胺复合膜的抗污染性能。采用后处理工艺,对现有正渗透聚酰胺复合膜的活性层(即聚酰胺层)进行改性,通过多巴胺交联技术和原子转移自由基聚合反应,在正渗透聚酰胺复合膜表面接枝具有高亲水性的两性离子聚合物,从而提高正渗透聚酰胺复合膜的亲水性,降低电荷性和粗糙度,降低表面羧基浓度,有效提高膜的抗污染性能。
本实施方式采用原子转移自由基聚合反应ATRP,其发展较为成熟,所用化学试剂常见,同时,选用的商品化的两性离子,价格相对便宜,同时,采用的是ARGET-ATRP(电子转移活化再生催化剂原子转移自由基聚合),其特点是对氧气有一定的容忍度,需要的催化剂量少,便于以后大规模工业化开发。
本实施方式得到的抗污染正渗透聚酰胺复合膜在大幅度提高抗污染性能的同时,可保持其过水性能和对盐的选择性,另外,由于ATRP高度的可控性,可通过调节聚合物的生长时间等反应参数,对膜的传质和抗污染表面性能进行进一步调控和优化。
由于本实施方式首先借助多巴胺将引发剂接枝在TFC膜上,而多巴胺能在溶解氧的作用下发生氧化-交联反应,形成强力附着于固体材料表面的聚合多巴胺,即多巴胺除了TFC膜的聚酰胺,也可附着在各种不同的表面,因此本实施方式中的方法不仅适用于TFC膜,也同时适用于其他表面的抗污染改性。
具体实施方式二:本实施方式与具体实施方式一的不同点是:步骤一中将盐酸多巴胺溶解于二甲基甲酰胺中,然后通入氮气,在氮气保护下加入2-溴异丁酰溴和三乙胺,并在氮气保护下搅拌反应3h,得到耦合溶液。其他与具体实施方式一相同。
具体实施方式三:本实施方式与具体实施方式一或二之一不同点是:步骤一中所述的盐酸多巴胺的质量与二甲基甲酰胺的体积比为800mg:40mL。其他与具体实施方式一或二相同。
具体实施方式四:本实施方式与具体实施方式一至三之一不同点是:步骤一中所述的盐酸多巴胺的质量与2-溴异丁酰溴的体积比为800mg:0.26mL。其他与具体实施方式一至三相同。
具体实施方式五:本实施方式与具体实施方式一至四之一不同点是:步骤一中所述的盐酸多巴胺的质量与三乙胺的体积比为800mg:0.3mL。其他与具体实施方式一至四相同。
具体实施方式六:本实施方式与具体实施方式一至五之一不同点是:步骤二中的所述的耦合溶液与三羟甲基氨基甲烷缓冲液的体积为40mL:200mL。其他与具体实施方式一至五相同。
具体实施方式七:本实施方式与具体实施方式一至六之一不同点是:步骤二中向耦合溶液中加入三羟甲基氨基甲烷缓冲液,得到混合溶液,然后将正渗透聚酰胺复合膜表面的活性层浸没在混合溶液中,且保证正渗透聚酰胺复合膜表面的支撑层不与混合溶液接触,浸没时间为15min,得到接枝引发剂正渗透聚酰胺复合膜。其他与具体实施方式一至六相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同点是:步骤二中向耦合溶液中加入三羟甲基氨基甲烷缓冲液,得到混合溶液,然后将正渗透聚酰胺复合膜表面的活性层浸没在混合溶液中,且保证正渗透聚酰胺复合膜表面的支撑层不与混合溶液接触,浸没时间为30min,得到接枝引发剂正渗透聚酰胺复合膜。其他与具体实施方式一至七相同。
具体实施方式九:本实施方式与具体实施方式一至八之一不同点是:步骤三中将两性离子单体溶于异丙醇水溶液中,然后通入氮气,在氮气保护下加入氯化铜催化复合物,得到含两性离子单体溶液,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入抗坏血酸溶液,并在氮气保护下聚合反应1.5h~2h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜。其他与具体实施方式一至八相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同点是:步骤三中将两性离子单体溶于异丙醇水溶液中,然后通入氮气,在氮气保护下加入氯化铜催化复合物,得到含两性离子单体溶液,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入抗坏血酸溶液,并在氮气保护下聚合反应1.5h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜。其他与具体实施方式一至九相同。
具体实施方式十一:本实施方式与具体实施方式一至十之一不同点是:步骤三中将两性离子单体溶于异丙醇水溶液中,然后通入氮气,在氮气保护下加入氯化铜催化复合物,得到含两性离子单体溶液,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入抗坏血酸溶液,并在氮气保护下聚合反应2h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜。其他与具体实施方式一至十相同。
具体实施方式十二:本实施方式与具体实施方式一至十一之一不同点是:步骤三中所述的两性离子单体的质量与异丙醇水溶液的体积比为15.64g:200mL。其他与具体实施方式一至十一相同。
具体实施方式十三:本实施方式与具体实施方式一至十二之一不同点是:步骤三中所述的两性离子单体的质量与氯化铜催化复合物的体积比为15.64g:20mL。其他与具体实施方式一至十二相同。
具体实施方式十四:本实施方式与具体实施方式一至十三之一不同点是:步骤三中所述的两性离子单体的质量与抗坏血酸溶液的体积比为15.64g:12mL。其他与具体实施方式一至十三相同。
具体实施方式十五:本实施方式与具体实施方式一至十四之一不同点是:步骤三中所述的两性离子单体为磺基甜菜碱丙烯酸甲酯单体。其他与具体实施方式一至十四相同。
具体实施方式十六:本实施方式与具体实施方式一至十五之一不同点是:步骤三中所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成。其他与具体实施方式一至十五相同。
具体实施方式十七:本实施方式与具体实施方式一至十六之一不同点是:步骤三中所述的氯化铜催化复合物按以下步骤制备的:
将氯化铜和三丙二醇甲醚醋酸酯溶于异丙醇水溶液中,即得到氯化铜催化复合物;所述的氯化铜与三丙二醇甲醚醋酸酯的质量比为0.01:0.14;所述的所述的氯化铜的质量与异丙醇水溶液的体积比为0.01g:20mL;所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成。
其他与具体实施方式一至十六相同。
具体实施方式十八:本实施方式与具体实施方式一至十七之一不同点是:步骤三中所述的抗坏血酸溶液按以下步骤制备的:将抗坏血酸溶于异丙醇水溶液中,即得到抗坏血酸溶液;所述的抗坏血酸的质量与异丙醇水溶液的体积比为1g:10mL;所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成。其他与具体实施方式一至十七相同。
本发明内容不仅限于上述各实施方式的内容,其中一个或几个具体实施方式的组合同样也可以实现发明的目的。
采用下述试验验证本发明效果:
实施例1:结合图1,一种后处理制备抗污染正渗透聚酰胺复合膜的方法,具体是按以下步骤完成的:
一、耦合:将800mg盐酸多巴胺溶解于40mL二甲基甲酰胺中,然后通入氮气,在氮气保护下加入0.26mL 2-溴异丁酰溴和0.3mL三乙胺,并在氮气保护下搅拌反应3h,得到耦合溶液;
二、接枝引发剂:向40mL耦合溶液中加入200mL三羟甲基氨基甲烷缓冲液,得到混合溶液,然后将正渗透聚酰胺复合膜表面的活性层浸没在混合溶液中,且保证正渗透聚酰胺复合膜表面的支撑层不与混合溶液接触,浸没时间为30min,得到接枝引发剂正渗透聚酰胺复合膜;
三、接枝两性离子聚合物:将15.64g两性离子单体溶于200mL异丙醇水溶液中,然后在氮气保护下加入20mL氯化铜催化复合物,得到含两性离子单体溶液,然后通入氮气,在氮气保护下将接枝引发剂正渗透聚酰胺复合膜浸入含两性离子单体溶液中,然后在氮气保护下加入12mL抗坏血酸溶液,并在氮气保护下聚合反应1h,然后暴露于空气中终止聚合反应,取出后得到抗污染正渗透聚酰胺复合膜;
步骤三中所述的两性离子单体为磺基甜菜碱丙烯酸甲酯单体;
步骤三中所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成;
步骤三中所述的氯化铜催化复合物按以下步骤制备的:
将0.01g氯化铜和0.14g三丙二醇甲醚醋酸酯溶于20mL异丙醇水溶液中,即得到氯化铜催化复合物;所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成;
步骤三中所述的抗坏血酸溶液按以下步骤制备的:将1g抗坏血酸溶于10mL异丙醇水溶液中,即得到抗坏血酸溶液;所述的异丙醇水溶液由异丙醇和去离子水按异丙醇与去离子水体积比为1:1混合而成。
本实施例步骤二中所述三羟甲基氨基甲烷缓冲液的pH为8.5。
本实施例步骤二中所述的正渗透聚酰胺复合膜为高选择性正渗透聚酰胺复合膜,具体是按中国已公开专利“一种高选择性正渗透聚酰胺复合膜的原位制备方法”(申请号:201410815730.7)提供方法制备的。
图1是实施例1操作流程示意图;在步骤一中,在氮气保护下,首先将原子转移自由基聚合反应将要使用的引发剂耦合接枝到多巴胺上;在步骤二中,在空气和缓冲溶液的环境下使多巴胺接触正渗透聚酰胺复合膜表面,通过多巴胺自生形成聚合多巴胺的聚合反应,使引发剂接枝在正渗透聚酰胺复合膜表面;在步骤三中,主要依靠原子转移自由基聚合反应,使两性离子单体从引发剂的位置在正渗透聚酰胺复合膜上生长,通过改变反应时间,可调控接枝的两性离子聚合物改性层。
对实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜进行有机污染,所述的正渗透聚酰胺复合膜为步骤二中所述的正渗透聚酰胺复合膜;采用正渗透FO错流过滤模式,以天然有机物(NOM)、海藻酸钠和牛血清白蛋白(BSA)的混合物作为典型有机污染物对膜进行污染,直到搜集500mL的渗透液为止,监测两种膜在有机污染过程中的通量衰减趋势,如图2所示,图2是实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜污染过程中纯水通量衰减示意图,图中○实施例1得到的抗污染正渗透聚酰胺复合膜污染过程中纯水通量衰减曲线,图中□正渗透聚酰胺复合膜污染过程中纯水通量衰减曲线;由图2可知,经过实施例1改性后得到的抗污染正渗透聚酰胺复合膜在正渗透膜过滤过程中水通量下降趋势变慢,极大提高了膜的抗污染性能。
为了研究膜的传质性能参数的变化,采用了正渗透四部法对实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜进行了表征,所述的正渗透聚酰胺复合膜为步骤二中所述的正渗透聚酰胺复合膜;如图3所示,图3是水渗透系数-结盐渗透系数-构特性参数柱形图,图中A表示正渗透聚酰胺复合膜的水渗透系数柱形图,图中B表示正渗透聚酰胺复合膜的盐渗透系数柱形图,图中S表示正渗透聚酰胺复合膜的结构特性参数柱形图,图中a表示实施例1得到的抗污染正渗透聚酰胺复合膜的水渗透系数柱形图,图中b表示实施例1得到的抗污染正渗透聚酰胺复合膜的盐渗透系数柱形图,图中s表示实施例1得到的抗污染正渗透聚酰胺复合膜的结构特性参数柱形图,从图3可以看出,实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜的结构特性参数(S)很相近,表明改性并不改变膜材料支撑层的传质性能。这主要是由于改性是在正渗透聚酰胺复合膜的活性层表面,不会对支撑层的厚度、孔隙率、孔道曲折度等结构特性产生直接影响。表面接枝两性离子聚合物改性主要影响活性层的传质性能,实施例1得到的抗污染正渗透聚酰胺复合膜与正渗透聚酰胺复合膜对比可知,水渗透系数(A)发生了轻微的降低,盐渗透系数(B)有轻微的升高,但是都不显著,表明本发明原位接枝两性离子聚合物改性并不会破坏正渗透聚酰胺复合膜的完整性和选择性。
- 还没有人留言评论。精彩留言会获得点赞!