一种气相色谱法检测甲磺酸仑伐替尼中甲磺酸酯类基因毒性杂质的方法与流程
1.本发明涉及药物分析领域,特别涉及一种抗肿瘤药物中可能含有的甲磺酸酯类基因毒性杂质检测的气相色谱法。
背景技术:
2.甲磺酸仑伐替尼是一种酪氨酸激酶(rtk)受体抑制剂,可抑制血管内皮生长因子(vegf)受体vegfr1(flt1)、vegfr2(kdr)和vegfr3(flt4)的激酶活性,另外还可抑制其它促血管生成和肿瘤发生通路相关的rtk,包括成纤维细胞生长因子(fgf),受体fgfr1、2、3和4,血小板衍生生长因子(pdgf)受体pdgfrα、kit和ret。
3.甲磺酸仑伐替尼结构式为:
[0004][0005]
甲磺酸酯类物质是在甲磺酸仑伐替尼的生产工艺过程中可能产生的杂质,这类杂质具有基因毒性警示结构,根据ich m7指导原则,甲磺酸酯类物质需严格控制其含量。甲磺酸酯类物质在水相体系中易发生水解,热不稳定,在检测过程中灵敏度低,准确定量困难。
[0006]
在甲磺酸酯类物质的质量控制中,现有文献资料中国抗生素杂质2017年6月第42卷第6期公开的“gc
‑
ms法测定甲磺酸培氟沙星中的甲磺酸酯杂质的研究”中,报道了一种gc
‑
ms的方法检测甲磺酸酯类物质。该方法需要采用气相质谱联用仪,检测成本高,不利于实验室推广。现有文献药品鉴定2013年9月第3卷第17期“衍生气相色谱法测定甲磺酸伊马替尼中甲磺酸烷基酯类”中,报道了一种气相方法测定甲磺酸酯类物质的方法。该方法为峰面积外标法,当供试品为甲磺酸仑伐替尼时,供试品本身在衍生化过程中的干扰很大,回收率低于80%,方法准确度低,无法准确控制甲磺酸酯类物质。
[0007]
甲磺酸仑伐替尼是一种多靶点激酶抑制剂,用于治疗甲状腺癌、晚期肾细胞癌和肝癌。本品合成路线中可能存在的甲磺酸酯类物质具有基因毒性警示结构。这类杂质的限度控制需根据本品用药周期不超过10年,从严按终身用药计算。按ich m7指导原则要求,1.5μg/天
÷
最大日服用剂量24mg/天=62.5ppm。因此,为有效控制样品质量,需要找到一种适合在常用气相色谱仪上检测甲磺酸酯类物质的方法。
技术实现要素:
[0008]
为克服现有方法不足,本发明提供了一种气相色谱法检测甲磺酸仑伐替尼中的甲磺酸酯类基因毒性杂质的方法,该方法通过在实验室常见的气相色谱仪上检测甲磺酸酯类物质,并且通过标准加入法能有效扣除供试品本身带来的检测干扰,方法检测成本和准确
度方面有明显优势。
[0009]
为了达到上述目的,本发明提供如下技术方案:
[0010]
一种气相色谱法检测甲磺酸仑伐替尼中甲磺酸酯类基因毒性杂质的方法,包括以下步骤:
[0011]
(1)称取碘化钠和维生素c,加水溶解并稀释,得到衍生化溶液;
[0012]
(2)称取甲磺酸仑伐替尼供试品,置于顶空瓶中,加入溶剂水浴60℃~100℃溶解,再加入步骤(1)中的衍生化溶液,进行衍生化反应,得到供试品溶液;
[0013]
(3)制备对照品溶液和供试品加标溶液;
[0014]
(4)采用顶空进样气相色谱法测定供试品溶液和供试品加标限度溶液中甲磺酸酯类基因毒性杂质,采用标准加入法计算甲磺酸仑伐替尼中甲磺酸酯类基因毒性杂质的含量;
[0015]
所述溶剂为乙腈与水的混合液,所述乙腈与水的体积比为60:40;所述甲磺酸酯类基因毒性杂质包括甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯;所述衍生化反应为使用衍生化溶液将目标物甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯衍生化为碘甲烷、碘乙烷和碘异丙醇。
[0016]
进一步地,所述步骤(1)中碘化钠与水的质量体积比为0.8~1.2g/ml;所述维生素c与水的质量体积比为0.0005~0.0015g/ml。
[0017]
进一步地,所述步骤(2)中溶剂与衍生化溶液的体积比1:6~3:1;所述甲磺酸仑伐替尼供试品与溶剂的质量体积比为20~60mg/ml。
[0018]
进一步地,所述步骤(3)中对照品溶液包括对照品储备液、50%对照品溶液、100%对照品溶液和150%对照品溶液;所述供试品加标溶液包括供试品加标50%限度溶液、供试品
[0019]
加标100%限度溶液和供试品加标150%限度溶液,制备步骤为:
[0020]
(1)对照品储备液的制备:称取甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯,加溶剂定量稀释,得到对照品储备液,所述甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯分别与对照品储备液的质量体积比为25~500μg/ml;
[0021]
(2)50%对照品溶液的制备:移取对照品储备液,加溶剂定量稀释,得到50%对照品溶液,所述甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯分别与50%对照品溶液的质量体积比为2.5μg/ml;
[0022]
(3)100%对照品溶液的制备:移取对照品储备液,加溶剂定量稀释,得到100%对照品溶液,所述甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯分别与100%对照品溶液的质量体积比为5μg/ml;
[0023]
(4)150%对照品溶液的制备:移取对照品储备液,加溶剂定量稀释,得到150%对照品溶液,所述甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯分别与150%对照品溶液的质量体积比为7.5μg/ml;
[0024]
(5)供试品加标50%限度溶液的制备:取甲磺酸仑伐替尼供试品0.2g,置顶空瓶中,加50%对照品溶液2.0ml,水浴溶解后加衍生化溶液3.0ml,密封,得到供试品加标50%限度溶液;
[0025]
(6)供试品加标100%限度溶液的制备:取甲磺酸仑伐替尼供试品0.2g,置顶空瓶
中,加100%对照品溶液2.0ml,水浴溶解后加衍生化溶液3.0ml,密封,得到供试品加标100%限度溶液;
[0026]
(7)供试品加标150%限度溶液的制备:取甲磺酸仑伐替尼供试品0.2g,置顶空瓶中,加150%对照品溶液2.0ml,水浴溶解后加衍生化溶液3.0ml,密封,得到供试品加标150%限度溶液。
[0027]
所述步骤(1)至步骤(4)中溶剂为乙腈与水的混合液,所述乙腈与水的体积比为60:40;所述步骤(5)至步骤(7)中水浴温度为60℃~100℃
[0028]
进一步地,所述步骤(4)中甲磺酸酯类基因毒性杂质含量的计算方法为标准加入法;
[0029]
进一步地,所述标准加入法的计算步骤为:取供试品溶液、供试品溶液加标50%限度溶液、供试品加标100%限度溶液、供试品加标150%限度溶液分别顶空进样气相色谱检测,记录色谱图,碘甲烷、碘乙烷、碘异丙烷依次出峰,以各组分对照品加入含量(μg/ml)为x轴,以峰面积为y轴,绘制标准曲线;按以下公式计算供试品中各组分的含量;
[0030]
甲磺酸甲酯或甲磺酸乙酯或甲磺酸异丙酯(%)=|a|
×
10
‑3×
5/200
×
100;
[0031]
其中,a为标准曲线在x轴的截距;200为供试品取样量,mg。
[0032]
进一步地,所述步骤(4)中采用气相色谱法的进样方法为顶空进样法,测定条件为:
[0033]
顶空平衡温度为70~130℃;
[0034]
顶空平衡时间为15~45min;
[0035]
色谱柱为毛细管色谱柱;
[0036]
柱长为30m,直径为0.32mm,膜厚为1.8μm;
[0037]
柱温:采用程序升温,初始柱温为35~45℃,维持5分钟;升温速率为10~30℃/min,升至200℃,维持10分钟;
[0038]
进样口温度为180~220℃;
[0039]
检测口温度为210~250℃;
[0040]
柱压力为40~60kpa;
[0041]
流速为2
‑
5ml/min;
[0042]
分流比为5:1~20:1;
[0043]
进样量为1ml;
[0044]
所述气相色谱法检测器为氢火焰离子化检测器。
[0045]
进一步地,所述毛细管色谱柱为中等极性、极性或适用于分离碱性物质的毛细管柱中的一种。
[0046]
进一步地,所述毛细管色谱柱为aglient db
‑
wax、agilent cp
‑
volamine和aglient db
‑
624中的一种;优选的毛细管色谱柱为aglient db
‑
624。
[0047]
与现有技术相比,本发明具有如下优点和有益效果:
[0048]
(1)本发明采用标准加入法,能有效扣除供试品本身带来的检测干扰,显著减少了主成分的基质干扰,能准确对甲磺酸酯类杂质进行定量分析,结果准确可靠,具有良好的灵敏度、精密度和准确度,尤其是准确度方面有明显优势;
[0049]
(2)本发明提供的方法,可以仅通过气相色谱法对甲磺酸酯类基毒杂质进行检测,
操作性强,降低了检测成本,方便在一般实验室推广应用。
附图说明
[0050]
图1为甲磺酸甲酯(碘甲烷)定位的气相色谱图;
[0051]
图2为甲磺酸乙酯(碘乙烷)定位的气相色谱图;
[0052]
图3为甲磺酸异丙酯(碘异丙烷)定位的气相色谱图;
[0053]
图4为供试品溶液的气相色谱图;
[0054]
图5为供试品加标50%限度溶液的气相色谱图;
[0055]
图6为供试品加标100%限度溶液的气相色谱图;
[0056]
图7为供试品加标150%限度溶液的气相色谱图;
[0057]
图8甲磺酸甲酯(碘甲烷)标准曲线;
[0058]
图9甲磺酸乙酯(碘乙烷)标准曲线;
[0059]
图10甲磺酸异丙酯(碘异丙烷)标准曲线。
具体实施方式
[0060]
以下将结合具体实施例对本发明提供的技术方案进行详细说明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
[0061]
实施例1
[0062]
1)试验溶液配制:
[0063]
(1)溶剂:乙腈与水按体积比60:40进行混合;
[0064]
(2)衍生化溶液:称取20g碘化钠和20mg维生素c,精密称定,加水溶解并稀释至20ml,得到每1ml中约含碘化钠1g、维生素c1mg的衍生化溶液。
[0065]
(3)空白溶液:称取10g碘化钠和10mg维生素c,精密称定,加水溶解并稀释至10ml,得到每1ml溶液中含碘化钠1g、维生素c1mg的空白溶液。
[0066]
(4)对照品储备液:分别称取25mg甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯,精密称定,加100ml溶剂定量稀释制成每1ml中约含甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯各250μg的溶液,作为对照品储备液。
[0067]
(5)50%对照品溶液:精密移取1ml对照品储备液,加100ml溶剂定量稀释制成每1ml中约含甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯各2.5μg的溶液,作为50%对照品溶液。
[0068]
(6)100%对照品溶液:精密移取2ml对照品储备液,加100ml溶剂定量稀释制成每1ml中约含甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯各5μg的溶液,作为100%对照品溶液。
[0069]
(7)150%对照品溶液:精密移取3ml对照品储备液,加100ml溶剂定量稀释制成每1ml中约含甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯各7.5μg的溶液,作为150%对照品溶液。
[0070]
(8)供试品溶液:取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加溶剂2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为供试品溶液。
[0071]
(9)供试品加标50%限度溶液:取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加50%对照品溶液2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为供试品加标100%限度溶液。
[0072]
(10)供试品加标100%限度溶液:取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加100%对照品溶液2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为供试品加标100%限度溶液。
[0073]
(11)供试品加标150%限度溶液:取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加150%对照品溶液2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为供试品加标150%限度溶液。
[0074]
(12)定位溶液的配制:分别称取甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯,精密称定,置于不同的量瓶中,加溶剂溶解并定量稀释至每1ml含各组分约0.25mg的溶液,作为定位贮备液;精密量取各定位贮备液1ml,分别置于不同顶空瓶中,分别加入衍生化溶液3ml,加溶剂1ml,密封。
[0075]
2)专属性测试
[0076]
顶空进样气相色谱测定条件为:
[0077]
仪器:安捷伦7890b气相色谱仪;7697a顶空自动进样器;
[0078]
检测器:氢火焰离子化检测器;
[0079]
色谱柱:aglient db
‑
624毛细管柱,30m
×
0.32mm,1.8μm;
[0080]
载气:高纯氮气;
[0081]
进样口温度:200℃;检测口温度:230℃;
[0082]
柱压:50kpa;
[0083]
柱温:采用程序升温:40℃维持5分钟,20℃/min升至200℃,维持10分钟;
[0084]
顶空条件:顶空平衡时间30min;顶空平衡温度100℃;
[0085]
柱流速为3ml/min;
[0086]
分流比为10:1;
[0087]
进样量为1ml
[0088]
专属性测定方法:分别取定位溶液、供试品溶液、供试品加标100%限度溶液、空白溶剂顶空进样,记录色谱图。
[0089]
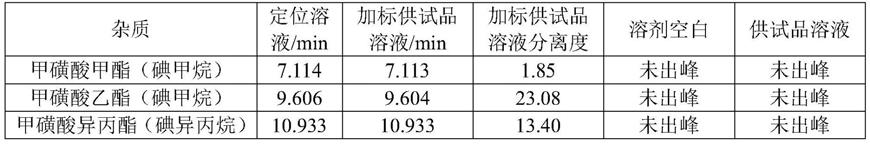
空白溶剂对测定无干扰,各化合物保留时间及分离度见下表。
[0090]
表1分析方法专属性结果
[0091][0092]
结论,本色谱条件下,甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯组分经衍生化后分别生成碘甲烷、碘乙烷和碘异丙烷,各组分均完全分离;溶剂及供试品溶液中无杂质峰干扰甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的检测,方法专属性良好。
[0093]
实施例2外标法对比验证
[0094]
取溶液的配制项下,溶剂、100%对照品溶液、供试品加标50%限度溶液、供试品加标100%限度溶液、供试品加标150%限度溶液按专属性项下色谱条件顶空进样,以峰面积外标法测定回收率。
[0095]
表2外标法回收率结果
[0096][0097]
结论,本色谱条件下,甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯组分经衍生化后分别生成碘甲烷、碘乙烷和碘异丙烷,各组分回收率均不在80%~120%范围内,方法准确度不符合要求。需进步优化方法排除供试品的干扰。
[0098]
实施例3标准加入法验证
[0099]
将供试品溶液、供试品溶液加标50%限度溶液、供试品加标100%限度溶液、供试品加标150%限度溶液分别顶空进样记录色谱图,如图4至图7,根据各色谱图,按标准加入法计算甲磺酸酯类杂质的含量。供试品加标溶液色谱图中碘甲烷(甲磺酸甲酯)、碘乙烷(甲磺酸乙酯)、碘异丙烷(甲磺酸异丙酯)依次出峰,各成分峰之间的分离度均大于1.5。以各甲磺酸酯类对照品加入含量(μg/ml)为x轴,以峰面积为y轴,绘制标准曲线。按以下公式计算供试品中各甲磺酸酯类的含量。
[0100]
甲磺酸甲酯或甲磺酸乙酯或甲磺酸异丙酯(%)=|a|
×
10
‑3×
5/200
×
100
[0101]
其中,a为标准曲线在x轴的截距;200为供试品取样量,mg。
[0102]
表3标准加入法试验结果
[0103][0104]
标准加入法验证对应的甲磺酸甲酯(碘甲烷)标准曲线、甲磺酸乙酯(碘乙烷)标准曲线、甲磺酸异丙酯(碘异丙烷)标准曲线见说明书附图8、图9和图10。
[0105]
结论:标准加入法能扣除供试品干扰,本方法线性关系良好,甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的浓度加入量与峰面积线性相关系数平方均大于0.995。标准加入法可用于样品中的甲磺酸酯类控制检测。
[0106]
实施例4耐用性试验
[0107]
其他条件同实施例1,不同的是,分别改变顶空平衡时间、顶空平衡温度、进样口温度、检测器温度、初始柱温、升温速率、柱压、衍生化溶液碘化钠加入量、衍生化溶液维生素c加入量以及供试品溶解温度,考察方法的耐用性。
[0108]
分别取实施例1制备的供试品溶液、供试品加标50%限度溶液、供试品加标100%限度溶液和供试品加标150%限度溶液,按表4~5的参数顶空进样气相色谱测定;结果如表
6所示。
[0109]
表4耐用性测定条件(aglient db
‑
624,毛细管柱30m
×
0.32mm,1.8μm)
[0110][0111]
表5耐用性测定条件(不同色谱柱类型)
[0112][0113]
表6耐用性测定条件下甲磺酸酯类杂质检测结果
[0114][0115]
以上结果表明,改变色谱参数,各耐用性条件与优选条件测得各组分含量差值均小于限度的20%(即为62.5*0.2=17ppm),甲磺酸甲酯与优选条件的最大差值为7.5ppm;甲磺酸乙酯与优选条件的最大差值为1.6ppm;甲磺酸异丙酯与优选条件的最大差值为1.2ppm。改变色谱柱类型对甲磺酸酯类的分离及检测结果均无显著影响。
[0116]
实施例5灵敏度试验
[0117]
顶空进样气相色谱检测条件同实施例4中的优选条件。
[0118]
取50%对照品溶液(每1ml中含甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各约2.5μg),加溶剂定量稀释制成每1ml中含甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各约0.25μg的溶液,作为检测限贮备液。
[0119]
取50%对照品溶液(每1ml中含甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各约2.5μg),加溶剂定量稀释制成每1ml中含甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各约1μg的溶液,作为定量限贮备液。
[0120]
取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加检测限贮备液2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为检测限溶液。
[0121]
取甲磺酸仑伐替尼供试品约0.2g,精密称定,置顶空瓶中,加定量限贮备液2.0ml,80℃水浴溶解后加衍生化溶液3.0ml,密封,作为定量限溶液。
[0122]
取上述定量限溶液各5份、检测限溶液各3份顶空进样,试验结果见表7至表9。
[0123]
表7甲磺酸甲酯检测限与定量限结果
[0124][0125]
表8甲磺酸乙酯检测限与定量限结果
[0126][0127]
表9甲磺酸异丙酯检测限与定量限结果
[0128][0129]
结论:甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的定量限浓度分别为1.03μg/ml、1.03μg/ml、0.96μg/ml,约相当于供试品浓度的26ppm、26ppm、24ppm;甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的检测限浓度分别为0.31μg/ml、0.31μg/ml、0.29μg/ml,约相当于供试品浓度的8ppm、8ppm、7ppm;即甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯含量分别大于8ppm、8ppm、7ppm即可被检出,含量分别大于26ppm、26ppm、24ppm,即可被准确定量。
[0130]
本发明方案所公开的技术手段不仅限于上述实施方式所公开的技术手段,还包括由以上技术特征任意组合所组成的技术方案。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1