一种三嗪类化合物、制备方法及其应用与流程
本发明涉及一种新化合物,具体涉及一种三嗪类化合物、制备方法及其应用。属于材料技术领域。
背景技术:
膨胀型无卤阻燃剂是一种含氮、磷的复合阻燃剂,主要用于聚烯烃的阻燃,是近年来发展最快的一类无卤阻燃剂。含膨胀型无卤阻燃剂的高分子材料受热燃烧时,会在聚合物表面形成一层均匀的泡沫炭质层,该炭质层在凝聚相起到隔热、隔氧、抑烟和防止熔滴的作用,可有效阻止有毒烟气和腐蚀性气体的产生和扩散,是一种非常有前途的新型阻燃剂。
一般而言,膨胀型阻燃剂包括三部分,即炭源(常为多羟基化合物,如季戊四醇)、酸源(如聚磷酸铵)及气源(如三聚氰胺)。成炭剂作为膨胀型阻燃体系中最重要的组分之一,其溶解度、热稳定性和成炭性在很大程度上决定了膨胀型阻燃剂的应用范围和阻燃效率。三嗪类大分子成炭剂是一种富含叔氮结构的化合物,有优良的发泡效果和成炭效果,并且具有无卤、低毒,分解温度高,对材料的物理机械性能影响小,抗渗出,阻燃效率高等优点,与传统炭源相比成炭量优异,不析出、耐迁移,水溶性低。
近年来,三嗪类大分子成炭剂的制备及其在阻燃领域应用的专利不断公开,中国科学院宁波材料技术与工程研究所公开了三嗪系超支化大分子成炭剂的制备方法(专利CN200910098421.1),采用一锅法将二氨基或二羟基化合物与非质子性有机溶剂混合,加入催化剂,在惰性气体中加入三聚氯氰的非质子性有机溶液和封端剂,制备三嗪系超支化大分子成炭剂;该研究所还公开了含芳香族链结构的三嗪成炭剂及其制备方法(专利CN200910099717.5),由结构通式为NH2-Ar-NH2,HO-Ar-OH或HS-Ar-SH的芳香族二元化合物和单取代三聚氯氰在催化剂作用下缩聚得到;上海化工研究院公开了一种以三聚氯氰和二元胺为反应产物制备三嗪类成炭剂及无卤膨胀型阻燃剂的方法(专利CN200710170519.4);广州大学公开了胺基三嗪衍生物大分子成炭剂及聚丙烯阻燃剂制备方法(专利CN201610509222.5),以三聚氯氰、含烷基胺的混合胺和缚酸剂为原料得到胺基三嗪衍生物大分子成炭剂。
以上制备方法皆存在有机溶剂用量大(主要为丙酮)、环境污染严重,后续处理困难,产品热稳定性不高,炭残留量低等缺点,并且反应周期普遍较长,在20小时以上。因此研究一种简单、高效、性能好的三嗪类衍生物成炭剂具有很好的应用价值。
技术实现要素:
本发明的目的是为克服上述现有技术的不足,提供一种三嗪类化合物。
本发明还提供了上述一种三嗪类化合物的制备方法及其应用。
为实现上述目的,本发明采用下述技术方案:
一种三嗪类化合物,其结构式如下:
上述一种三嗪类化合物的制备方法,包括步骤:
(1)在三乙胺的作用下,2,2'-二羟基联苯与三氯氧磷发生缩合反应,得到6-氯二苯丙[d,f][1,3,2]二氧杂磷杂环戊烷-6-氧化物,反应式如下:
(2)6-氯二苯丙[d,f][1,3,2]二氧杂磷杂环戊烷-6-氧化物发生水解反应,得到含有6-羟基二苯丙[d,f][1,3,2]二氧杂磷杂环戊烷-6-氧化物的反应液,接着与三聚氰胺发生成盐反应,即得产物,反应式如下:
优选的,步骤(1)的具体方法是:先将2,2'-二羟基联苯与三乙胺混合均匀,然后搅拌下缓慢滴加三氯氧磷,滴加完毕后调整温度至-10~20℃,反应4~7小时。
进一步优选的,使用甲苯作为反应溶剂,用于溶解反应物料和提供反应环境,甲苯与2,2'-二羟基联苯的质量比为2~3.5:1,更进一步优选为2.3:1。
进一步优选的,2,2'-二羟基联苯、三乙胺与三氯氧磷的摩尔比为1~1.2:1.1~1.3:1,进一步优选的1.05:1.1:1。
进一步优选的,三氯氧磷的滴加时间为1~3小时,更进一步优选为1.5小时。
进一步优选的,反应温度为10℃,反应时间为5小时。
优选的,步骤(2)的具体方法是:加入水和三聚氰胺,加热回流反应3~5小时,反应结束后冷却,过滤,干燥,即得产物。
进一步优选的,三聚氰胺与三氯氧磷的摩尔比为1:3~4,更进一步优选为1:3.2,水与三聚氰胺的质量比为800:37.9。
进一步优选的,步骤(1)中使用甲苯作为反应溶剂,在加入水和三聚氰胺之前,先旋蒸除去甲苯。
进一步优选的,反应时间为3.5小时。
进一步优选的,所述干燥为80℃真空干燥6~20小时。
上述的一种三嗪类化合物在单独使用或者作为膨胀型阻燃体系的成炭剂中的应用。
本发明的有益效果:
本发明以2,2'-二羟基联苯、三氯氧磷、三乙胺、三聚氰胺为主要原料,制得一种新型的三嗪类化合物,它不仅有炭源(联苯结构),而且还有酸源(磷酸结构)和气源(三聚氰胺),该成炭剂成炭性能好,热分解温度高,具有良好的稳定性,残炭量大。
本发明的制备方法,反应条件温和,副反应少,操作流程简单,反应周期大大缩短,所用原料安全、易得,以水为反应介质,避免了有机溶剂的使用,简化了后处理过程,便于工业化生产。本发明的溶剂及母液可回收套用,工艺操作流程简单,避免了现有工艺反应时间过长和有机溶剂用量大等缺点。
附图说明
图1是本发明实施例1所得三嗪类化合物的红外谱图。
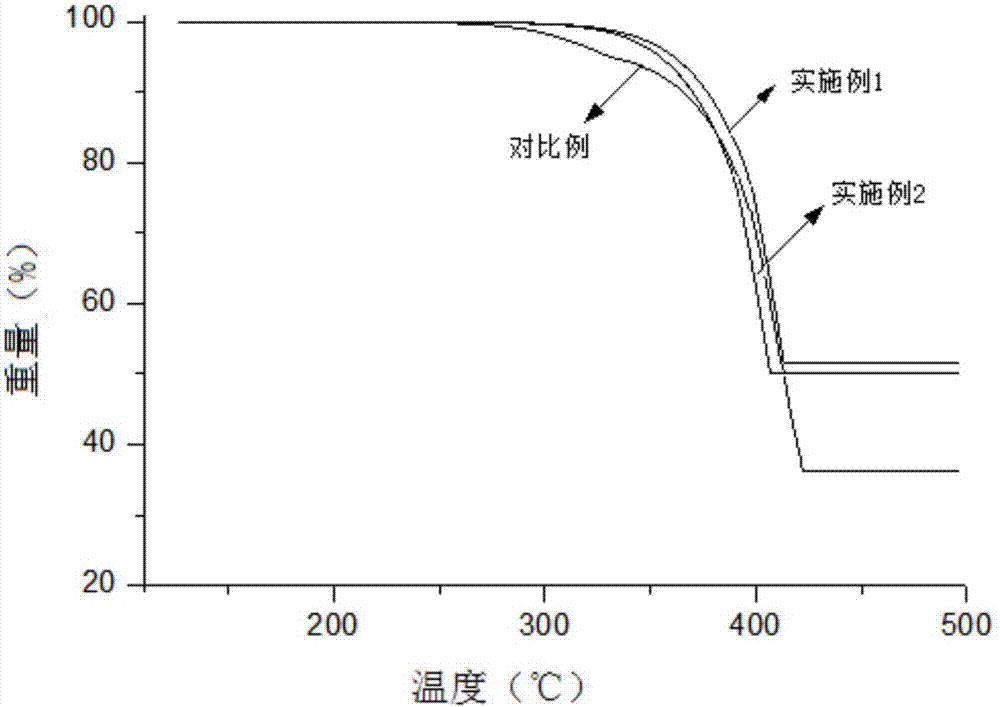
图2为实施例1、实施例2和对比例所得成炭剂的热重分析图。
具体实施方式
下面结合附图和实施例对本发明进行进一步的阐述,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
实施例1:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加188.0g(1mol)2,2'-二羟基联苯(99.0%),433.0g甲苯(99.5%),132.9g(1.3mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,1.0h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应4h。旋蒸甲苯,加入800g水,37.9g(0.3mol)三聚氰胺(99.8%),100℃回流反应3h后降温、过滤、烘干,得产物801.1g,以三氯氧磷计收率96.0%,热重分析500℃的成炭量为51.8%,热分解温度305℃。
图1是实施例1所得三嗪类化合物的红外谱图,从图1可以看出存在三聚氰胺基团(1650~1000cm-1)、联苯基团(3500~2800cm-1)和磷氧基团(600~750cm-1),与目标产物的结构吻合。
实施例2:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加225.6g(1.2mol)2,2'-二羟基联苯(99.0%),433.0g甲苯(99.5%),112.4g(1.1mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,1.5h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应5h。旋蒸甲苯,加入800g水,39.1g三聚氰胺(0.31mol)(99.8%),100℃回流反应3.5h后降温、过滤、烘干,得产物821.5g,以三氯氧磷计收率98.5%。热重分析500℃的成炭量为50.2%,热分解温度304℃。
实施例3:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加188.0g(1mol)2,2'-二羟基联苯(99.0%),433.0g甲苯(99.5%),102.2g(1mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,3h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应7h。旋蒸甲苯,加入800g水,37.9g(0.3mol)三聚氰胺(99.8%),100℃回流反应5h后降温、过滤、烘干,得产物809.8g,以三氯氧磷计收率97.1%。热重分析500℃的成炭量为50.2%,热分解温度304℃。
实施例4:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加197.0g(1.05)2,2'-二羟基联苯(99.0%),433.0g甲苯(99.5%),112.4g(1.1mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,1.5h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应5h。旋蒸甲苯,加入800g水,31.5g(0.25mol)三聚氰胺(99.8%),100℃回流反应3.5h后降温、过滤、烘干,得产物816.5g,以三氯氧磷计收率97.9%。
实施例5:
温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加188.0g(1mol)2,2'-二羟基联苯(99.0%),433.0g甲苯(99.5%),132.9g(1.3mol)三乙胺(含量99.0%),开启搅拌,控制温度在20℃,1.0h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应4h。旋蒸甲苯,加入800g水,37.9g(0.3mol)三聚氰胺(99.8%),100℃回流反应3h后降温、过滤、烘干,得产物806.5g,以三氯氧磷计收率96.7%。
实施例6:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加188.0g(1mol)2,2'-二羟基联苯(99.0%),658.0g甲苯(99.5%),132.9g(1.3mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,1.0h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应4h。旋蒸甲苯,加入800g水,37.9g(0.3mol)三聚氰胺(99.8%),100℃回流反应3h后降温、过滤、烘干,得产物803.1g,以三氯氧磷计收率96.3%。
实施例7:
向带有搅拌器、温度计和恒压滴液漏斗的2L四口圆底烧瓶中,加188.0g(1mol)2,2'-二羟基联苯(99.0%),376.0g甲苯(99.5%),132.9g(1.3mol)三乙胺(含量99.0%),开启搅拌,控制温度在10℃,1.0h滴加三氯氧磷155.6g(1mol)(98.5%),继续反应4h。旋蒸甲苯,加入800g水,37.9g(0.3mol)三聚氰胺(99.8%),100℃回流反应3h后降温、过滤、烘干,得产物786.5g,以三氯氧磷计收率94.3%。
通过实施例1~7的方法,反应条件更温和,收率提高,简化了后处理过程,便于工业化生产。
对比例:
一种由N-N-二甲基乙酰胺、三聚氯氰、二氨基二苯醚制备的芳香族链结构三嗪类成炭剂,热重分析500℃成炭量为38.5%,热分解温度285℃。
图2为实施例1、实施例2和对比例所得成炭剂的热重分析图。从图2看以看出,实施例1所得成炭剂热重分析500℃的成炭量为51.8%,热分解温度305℃;实施例2所得成炭剂热重分析500℃的成炭量为50.2%,热分解温度304℃;而对比例中成炭剂热重分析500℃成炭量为38.5%,热分解温度285℃。本发明所得成炭剂成炭性能好,残炭量大,热分解温度高,具有良好的稳定性。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!