基于凝血酶和凝血因子的双靶点抗凝活性物质的液相色谱‑质谱筛选方法与流程
本发明属于药物分析
技术领域:
,具体涉及基于凝血酶和凝血因子的双靶点抗凝活性物质的液相色谱-质谱筛选方法。
背景技术:
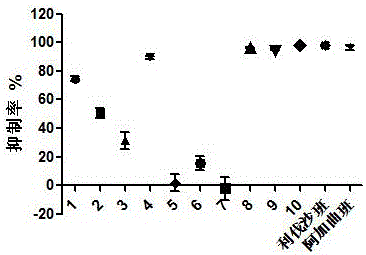
:酶是机体中重要的药物靶点之一,现有的药物近半数都是小分子酶抑制剂。运用酶靶点筛选抑制剂已有较多研究:有报道将文丘里氏管应用于质谱接口技术,开发了文丘里-常压超声喷雾离子源质谱系统,并将其用于高通量筛选血管紧张素转换酶的抑制剂(Liu,N.,etal.,AnalyticaChimicaActa,2016.913:86-93.);近几年,酶固定化技术和磁性纳米颗粒结合的方法开发报道也很多,例如采用酶固定化磁珠结合高效液相色谱-紫外检测技术用于筛选GSK-3β的抑制剂(Li,Y.,etal.,JournalofChromatographyA,2015.1425:8-16.)和从药用植物中快速发现α-淀粉酶抑制剂(Li,Y.,etal.,JournalofChromatographyB,2014.960:166-173.);另外,Kumar等人采用计算机辅助药物设计概念,利用3D形状匹配,静电势相似性评估和分子对接的方法,使用虚拟筛选和骨架转移法筛选具有SUMOE1抑制活性的新颖结构(Kumar,A.,etal.,Bioorganic&MedicinalChemistryLetters,2016.26(4):1218-1223.);血红素肽片段具有类似于过氧化酶的活性,在双氧水存在下可以氧化有机分子。基于这个原理,开发了用于胰蛋白酶活性测定和抑制剂筛选的荧光检测方法(Zhang,L.,etal.,Talanta,2016.161:535-540.);半胱胺封端的CdTe量子点(QDs),其表面的氨基会通过静电作用和氢键作用与加入的ATP相互作用,从而产生明显的荧光增强作用。有学者利用这个现象,开发了用于酸性磷酸酶(ACP)活性测试及其抑制剂的筛选(Wang,J.,etal.,SensorsandActuatorsB:Chemical,2016.234:470-477.);Morris团队开发荧光生物传感器探测细胞周期蛋白依赖性激酶,并监控了他们在体外和活细胞中含量和活性的变化,以发现新的用于癌症治疗的抑制剂(Prével,C.,etal.,EuropeanJournalofMedicinalChemistry,2014.88:74-88.);现今,将样品前处理、在线分离和分析等多种技术联合用于筛选酶抑制剂的报道屡见不鲜,既有利用新型样品制备和在线HPLC-DAD-MS/MS-BCD联合分析,快速筛选和表征中药提取物中的特异性α-葡萄糖苷酶抑制剂(LiD.Q.,etal.,JournalofPharmaceuticalandBiomedicalAnalysis2014.88:130–135);也有通过在线固相萃取和HPLC联用使用紫外检测器测定花生四烯酸,从而筛选细胞溶质磷脂酶A2α的抑制剂(Hanekamp,W.andM.Lehr,JournalofChromatographyB,2012.900:79-84.);还有通过组合HPLC-HRMS-SPE-NMR方法得到可食用海藻的高分辨率α-葡萄糖苷酶抑制谱,并鉴定了其中的α-葡萄糖苷酶的抑制剂(Liu,B.,etal.,FoodChemistry,2016.203:16-22.);Zhou等用超滤-高效液相色谱法对茯苓的不同溶剂提取部位进行了β-葡萄糖苷酶抑制活性的筛选,并对有活性的部位结合高速逆流色谱与SephadexLH-20色谱两种技术进行了进一步的分离纯化,得到了3种抑制活性的化合物,并对其进行了结构解析(Zhou,X.,etal.,JournalofChromatographyB,2015.985:149-154.)。近年来,新兴的纳米微球技术、酶固定化技术、荧光量子点成像技术、表面等离子共振技术和液相-质谱联用技术等被整合起来应用于酶抑制剂筛选,形成一系列新的高效率、高通量的筛选技术。目前公开的文献包括:采用UHPLC-Q-TOF/MS和磁珠偶联法筛选凝血酶抑制剂(Cao,J.,etal.,JournalofChromatographyA,2016.1468:86-94.);利用基于Au@GS和DNA-CoPdNPs双信号传导的超灵敏的电化学适配体传感器检测凝血酶酶活,并用于抑制剂的筛选(Wang,Y.,etal.,BiosensorsandBioelectronics,2016.80:640-646.);有文献基于多肽微阵列芯片,采用荧光和共振光散射法测定人血样本中凝血酶抑制剂的抑制活性(Su,M.,etal.,ChineseJournalofAnalyticalChemistry,2015.43(2):199-206.)。上述方法还都仅限于实验室水平的研究,尚未运用到实际生活中。2013年,多肽微阵列芯片在血浆中检测凝血酶活性及筛选凝血酶抑制剂的方法(CN201310282186.X)申请了专利,但该专利使用血浆样本,易受血浆本身特异性的干扰,且前期样品制备时间较长。目前常采用的抗凝药物的筛选技术是光学法和磁珠法。光学法通常包括:比浊法和光谱法(紫外光谱法和荧光光谱法)。比浊法包括散射比浊和透射比浊,相对检验的专一性不强,而且容易受到血浆特异性干扰。光谱法必须保证酶反应体系中有发光物质存在,且该物质的含量变化能灵敏地反应出酶反应的催化过程。现实中的天然底物很多都不具有发光能力,人们通过对其衍生化处理,使其具有发光基团,进而采用光谱法分析。这样本身就破坏了底物的天然状态,不能最真实的反映酶催化情况;其次,前处理相应增加了工作量,衍生化处理及分离等工作耗费大量人力、物力;再次,易受其他发光物质的干扰。在现实应用中,常需要和毛细管电泳和高效液相色谱联用。光谱法虽然可操作性强,准确度高,但其灵敏度低,不宜进行微量分析。磁珠法包括光电探测和电磁探测,利用磁珠在磁场运动产生电信号原理,分别借助光电探测和电信号监测的手段检测磁珠在磁场中的运动规律。该法虽然不受特异血浆的干扰,试剂用量相对少,但是,磁珠的重量、杯壁的光滑程度均对测量造成影响。技术实现要素:目前抗凝药物筛选方法存在易受特异血浆干扰、样品需要额外衍生和筛选速度慢,易出现假阳性的问题。本发明的目的在于克服以上不足,提供了一种高特异性、高灵敏度、高通量和方便快速的基于凝血酶和凝血因子的双靶点抗凝活性物质的液相色谱-质谱筛选方法。本发明建立了针对双酶为靶点筛选酶抑制剂的高效液相和质谱法联用方法,避免了样品前期的制备和衍生等不必要的步骤,自动化程度高,操作简单,易于推广。为实现上述发明目的,本发明采用以下技术方案予以实现:本发明提供了基于凝血酶和凝血因子的双靶点抗凝活性物质的液相色谱-质谱筛选方法,所述筛选方法包括如下步骤:(1)配制靶标酶酶反应产物的系列标准溶液,进行高效液相-质谱联用分析,建立靶标酶酶反应产物的定量方法;(2)将凝血酶和凝血因子及其对应的底物与待筛选化合物混合后,得到用于药物筛选的酶反应溶液,30-50℃下反应直至反应完成;去离子水为空白对照;(3)然后进行高效液相-质谱联用分析:测得所述靶标酶酶反应产物的峰面积,以抑制率为评价标准:抑制率=1-(待筛选化合物的质谱响应峰面积/空白对照的质谱响应峰面积),筛选出具有抗凝作用的活性物质。进一步的:所述步骤(1)中靶标酶酶反应产物为对硝基苯胺和7-胺基-4-甲基香豆素。进一步的:所述步骤(2)和(3)之间还设有步骤:对用于药物筛选的经酶反应后的溶液进行样品分离纯化。进一步的:所述分离纯化依次包括除蛋白和除盐步骤。进一步的:所述除蛋白步骤为:向所述酶反应后的溶液中加入与所述步骤(2)的酶反应溶液等体积的除蛋白剂,超速离心取上清。进一步的:使用苯磺酸基强阳离子交换型固相萃取柱对除蛋白后的溶液进行除盐处理。进一步的:所述步骤(1)和(3)中高效液相色谱的条件为:采用反向色谱柱;流动相为甲醇和水的混合液;流速为0.4-0.6mL/min;进样量为2-20μL;流动相洗脱时间为0.5-1.5分钟。进一步的:所述步骤(1)和(3)中质谱条件为电喷雾质谱正离子模式;碎裂电压:50-150V;干燥气温度:300-350℃;雾化器电压:10-20psi;干燥气流速:8-10L/min;毛细管电压:3500-4000V;定量方式为多重离子监测模式,采用的离子对为:m/z139-122和176-159,碰撞能量CE为8-20V。与现有技术相比,本发明的优点和技术效果为:操作简单、经济、快捷,可用商品化底物代替血浆,一方面避免了血浆的特异性差异的干扰;另一方面,也省去了血浆购买、保存等繁琐的工作。本发明利用了质谱的高特异性、高灵敏度和强稳定性,保证了所述筛选方法的干扰少、灵敏度高和重现性强;而且,质谱方法的自动化进样使得该筛选方法可以连续分析大量的样品,使该方法兼具高通量的特点。该筛选方法,可以同时针对两个靶点进行抗凝活性的筛选,对大批量样品库的筛选,更具实用性,且筛选出的活性物质作用靶点明确,为后续药物的开发奠定了理论基础。综上所述,本发明方法可以降低干扰、特异性高,操作简便、节约时间,有效的提高筛选效率和筛选成功率。附图说明图1为对硝基苯胺标准溶液的标准曲线。图2为实施例2中待筛选物质的抑制率结果。具体实施方式下面结合附图和具体实施例对本发明的技术方案做进一步详细的说明。实施例1本发明通过以下几个步骤获得了基于凝血酶和凝血因子Xa的双靶点抗凝活性物质的液相色谱-质谱筛选方法:1、采用外标法建立靶标酶酶反应产物-对硝基苯胺的定量方法利用底物S2238在凝血酶作用下,底物S2765在凝血因子Xa作用下都产生产物对硝基苯胺(pNA)的原理,使用对硝基苯胺作为定量标准。运用外标法,精确称取4.00mg的对硝基苯胺标准品,用50%乙腈水(v:v)溶液定容在100mL棕色容量瓶中,并作为母液保存在4℃冰箱里。分别取3mL、1mL、500μL、250μL、62.5μL、15μL和3μL母液用50%乙腈水(v:v)溶液定容在100mL的容量瓶中,配置成1.2μg·L-1、6μg·L-1、25μg·L-1、100μg·L-1、200μg·L-1、400μg·L-1和1200μg·L-1系列浓度的对硝基苯胺标准溶液,进行UHPLC-MS分析。仪器条件:高效液相的条件为:采用C18柱;流动相为40%H2O:60%CH3OH(V:V)等度洗脱1.2分钟;流速为0.4mL/min;进样量为2μL;柱温35℃。质谱条件为电喷雾质谱(ESI-MS)正离子模式;碎裂电压:90V;干燥气温度:350℃;雾化器电压:15psi;干燥气流速:10L/min;毛细管电压:4000V;定量方式为多重离子监测模式(MRM),采用的离子对为:母离子为m/z139,子离子为m/z122,碰撞能量(CE)为13V。以产物质谱峰面积为y轴和产物浓度为x轴,建立标准曲线。标准曲线如图1所示,线性回归方程为y=39.922x+65.101(R2=1)。2、对定量方法进行方法学验证对定量方法进行方法学验证,精密度、准确度、重复性、检测限和定量限、专属性和稳定性,方法均参照2015版药典四部《分析方法验证指导原则》。a.专属性试验:通过高分辨质谱测试出其分子量为139.0502,与计算分子量误差为0ppm。b.检测限和定量限:检测限:10ng·L-1(SNR=3.98±0.72;n=6);定量限:50ng·L-1(SNR=10.80±1.51;n=6);通过检测限和定量限结果可以看出,所述筛选方法检测灵敏度较高。c.精密度:选定该方法的定量下限0.05μg·L-1、中间浓度点25.00μg·L-1及高浓度点400μg·L-1,分别配制六份平行溶液,对每个浓度点采集的六个数据进行统计学分析,其相对标准偏差参照2015版药典四部《分析方法验证指导原则》,(1mg·L-1时,RSD%≤8;10μg·L-1时,RSD%≤15),结果如图1所示。相对标准偏差(RSD)都在可接受范围内,说明该定量方法的重复性较好。表1.方法学验证中精密度考察数据表峰面积0.05μg·L-1(n=6)25.00μg·L-1(n=6)400μg·L-1(n=6)Mean±SD29.80±1.791009.6±11.1418166.67±323.09RSD(%)6.001.101.78d.准确度:准确度用回收率衡量首先确定标准曲线的线性范围,结合酶反应检测出产物的最低浓度与最高浓度,选定10μg·L-1,100μg·L-1和625μg·L-1三个浓度。配制上述三个浓度80%、100%、120%水平的对硝基苯胺标准品溶液,每个浓度连续三针进样,得到其质谱响应信号。根据产物定量线性回归方程,将质谱响应峰面积代入其中,得出产物浓度,与配置的产物浓度的理论值进行比较,计算其回收率应符合2015版药典四部《分析方法验证指导原则》(1mg·L-1:回收率75%~120%;10μg·L-1:回收率70%~125%)。结果如表2所示。回收率的范围均在合理范围内,说明该定量方法的准确度高、重现性好。表2方法学验证中准确度考察数据表[pNA](μg·L-1)Recovery(%)Mean±SD(n=3)Recovery(%)RSD(%)(n=3)81.0037±0.108810.85101.0522±0.111410.59121.1343±0.08717.68800.9761±0.07497.671001.0513±0.01651.571201.0770±0.02011.865000.9991±0.03183.186251.0502±0.03403.477251.0919±0.04363.99e.稳定性:配制浓度为0.1mg·L-1的对硝基苯胺标准品,分别放置在4℃(低温离心)、25℃(常温放置)、37℃(酶反应)和85℃(浓缩)。每隔30分钟进行一次液质分析,根据质谱图中积分面积的变化,评价48小时内对硝基苯胺的稳定性。实验结果表明:48小时内,对硝基苯胺在4℃、25℃、37℃和85℃条件下均是稳定的,对定量结果无影响。3、酶反应体系的构建:分别精密称取10.00mg凝血酶的底物S2238和凝血因子Xa的底物S2765,转移至100mL容量瓶中,用去离子水稀释至刻度;分别精密称取10.00mg凝血酶和凝血因子Xa,转移至100mL容量瓶中,用pH=7.4的10mMTris-HCl缓冲液稀释至刻度;以上四个100mg·L-1的母液均保存在4℃冰箱里,供后续反应使用。分别取浓度为100mg·L-1的底物S2238、S2765各50μL与100μL去离子水混合,37℃下孵育;再加入凝血酶和凝血因子Xa母液各50μL构建用于双靶标酶酶抑制剂同时筛选的酶反应体系,37℃下反应15分钟。4、对所述酶反应体系进行分离纯化酶反应体系有维持酶活性的缓冲盐。但是非挥发性的缓冲盐不仅会影响质谱的灵敏度还会损伤仪器本身。所以好的除盐和除蛋白的前处理方法在质谱分析中极为重要。本发明采用有机溶剂沉淀法去除蛋白,上述酶反应体系,37℃下反应15分钟后,加入300μL的乙腈溶液淬灭酶反应体系。此处加入的乙腈既是反应淬灭剂,也是作为除蛋白步骤中的有机溶剂,12000rmp快速离心20分钟后获得除蛋白后的酶反应上清液。除盐采用SPE柱(强阳离子型苯磺酸基固相萃取柱),步骤如下:(1)活化:加入1mL的甲醇活化固相萃取柱的填充材料;(2)平衡:分别加入1mL的去离子水和1mL90%乙腈水溶液(v:v,并用乙酸调pH值为3.65-4)平衡固相萃取小柱;(3)上样:将100μL的上述除蛋白后的酶反应上清液加入固相萃取柱中,将柱中自然滤下的反应液收集在离心管中;(4)洗脱;加入1mL90%乙腈水溶液(v:v,并用乙酸调pH值为3.65-4)进行固相萃取柱匀速洗脱,速度控制在5mL·min-1,洗脱液也收集到离心管中;(5)浓缩:采用氮吹仪,控制金属浴温度85℃下加热,直至洗脱液被吹干;(6)复溶:加入100μL50%乙腈水(v:v)对吹干组分进行复溶,涡旋,离心(12000rmp,t=5min)取上清。5、高效液相-质谱联用分析将经过步骤4所述的除蛋白、除盐后的溶液按照如步骤1所述的方法和参数进行高效液相和质谱联用分析。实施例2本发明所述基于凝血酶和凝血因子的双靶点抗凝活性物质的液相色谱-质谱筛选方法具体步骤如下:1、配制靶标酶酶反应产物-对硝基苯胺的系列标准溶液,进行高效液相-质谱联用分析,建立靶标酶酶反应产物对硝基苯胺的定量方法如实施例1步骤1所述建立靶标酶酶反应产物对硝基苯胺的定量方法。对硝基苯胺的定量范围为1.20μg·L-1到1200μg·L-1。2、将双靶标酶及其底物与待筛选化合物混合得到用于筛选的酶反应溶液分别精密称取10.00mg的S2238、S2765,转移至100mL容量瓶中,用去离子水稀释至刻度。分别精密称取10.00mg的凝血酶和凝血因子Xa,转移至100mL容量瓶中,用pH=7.4的10mMTris-HCl缓冲液稀释至刻度;以上四个100mg·L-1的母液保存在4℃冰箱里,供后续实验使用。选取以下10种化合物:1.LPGS、2.LPMS-H、3.PGGS(L)、4.PGG、5.CS-2S、6.CS-3S、7.CS-1S、8.PGS、9.过硫化低分子量的CS、10.过硫化的CS二糖,从中筛选具有抗凝活性的物质。阿加曲班为凝血酶的直接抑制剂,作为凝血酶抑制作用的阳性对照;利伐沙班是凝血因子Xa的直接抑制剂,用作凝血因子Xa抑制作用的阳性对照。分别精密称取5.00mg的上述10种待筛选化合物和阳性对照药物,使用去离子水将其定容在50mL的容量瓶中,作为母液,保存在4℃冰箱中,供后续实验使用。分别取1mL待筛选化合物和阳性对照药物的母液,使用去离子水定容到10mL的容量瓶中,得到10mg·L-1的水溶液,作为待测抗凝物质筛选。将所述两种酶(凝血酶和凝血因子Xa)、两种底物(S2238和S2765)分别取50μL混合均匀,再加入100μL所述待筛选化合物(或阳性对照,或水),混合形成筛选双酶抑制剂的酶反应溶液,37℃下反应15分钟。3、对所述酶反应后的溶液进行除盐、除蛋白除蛋白操作:向酶反应后的溶液中加入与所述酶反应溶液等体积的乙腈(300μL),12000rmp快速离心20分钟后获得除蛋白后的酶反应上清液。除盐采用SPE柱(强阳离子型苯磺酸基固相萃取柱),步骤如下:(1)活化:加入1mL的甲醇活化固相萃取柱的填充材料;(2)平衡:分别加入1mL的去离子水和1mL90%乙腈水溶液(v:v,并用乙酸调pH值为3.65-4)平衡固相萃取小柱;(3)上样:将100μL的所述除蛋白后的酶反应上清液加入固相萃取柱中,将柱中自然滤下的反应液收集在离心管中;(4)洗脱;加入1mL90%乙腈水溶液(v:v,并用乙酸调pH值为3.65-4)进行固相萃取柱匀速洗脱,速度控制在5mL·min-1,洗脱液也收集到离心管中;(5)浓缩:采用氮吹仪,控制金属浴温度85℃下加热,直至洗脱液被吹干;(6)复溶:加入100μL50%乙腈水(v:v)对吹干组分进行复溶,涡旋,离心(12000rmp,t=5min)取上清。4、进行高效液相-质谱联用分析将上述除蛋白和除盐处理后的酶反应上清液进行高效液相-质谱联用分析,具体参数如下:高效液相的条件为:采用C18柱;流动相为40%H2O:60%CH3OH(v:v)等度洗脱1.2分钟;流速为0.4mL/min;进样量为2μL;柱温35℃。质谱条件为电喷雾质谱(ESI-MS)正离子模式;碎裂电压:90V;干燥气温度:350℃;雾化器电压:15psi;干燥气流速:10L/min;毛细管电压:4000V;定量方式为多重离子监测模式(MRM),采用的离子对为:母离子为m/z139,子离子为m/z122,碰撞能量(CE)为13V。测得产物对硝基苯胺峰面积,和添加阳性对照组标准凝血酶抑制剂(阿加曲班)和凝血因子Xa抑制剂(利伐沙班)的体系对比,并设立空白对照组(用去离子水代替抑制剂,其他条件都相同)。以抑制率为评价标准:抑制率=1-(待筛选样品的质谱响应峰面积/空白对照的质谱响应峰面积),筛选出具有抗凝作用的活性物质。计算待筛选物质的抑制率,整理绘制得图2。从实验结果中可以看出,10个化合物中有8个具有不同程度的抗凝活性,其中有4个化合物的抑制活性和商品化抑制剂相当,即获得具有双靶点抗凝活性的物质。本发明将外源性凝血的两个重要的靶标酶建立在一个反应体系中,将高效液相和质谱仪联用用于筛选抗凝药物,利用高效液相的分离作用对酶反应体系进行分离,采用质谱检测器对产物的产生量进行检测。可以快速准确地同时筛选出对两种靶标酶具有抑制活性的药物。以上实施例仅用以说明本发明的技术方案,而非对其进行限制;尽管参照前述实施例对本发明进行了详细的说明,对于本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的精神和范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1