一种光催化α‑二酮化合物的制备方法与流程
本发明属于医药化工中间体及相关化学技术领域,涉及到一种光催化α-二酮化合物的制备方法。
背景技术:
α-二酮类化合物是合成咪唑类、喹喔啉和吲哚酮氮氧化合物等重要的精细化学品的重要中间体,同时其本身就普遍存在于许多天然产物和生物活性分子。[Maurya R.,Singh R.,Deepak M.,et al.Phytochemistry 2004,65,915–920]。因此,开发高效、合成α-二酮类化合物的方法,具有重要的意义和应用价值。目前,合成α-二酮类化合物的方法主要有:亲核加成-消除法、偶联法以及氧化法等三种方法。其中,亲核加成-消除法发展较早,主要是利用格氏试剂、有机锂、有机锡试剂等和羧酸衍生物发生亲核加成—消去反应。但是,格氏试剂及有机锂试剂非常活泼,反应一般要在低温下进行且底物官能团适用性较差;另外,有机锡试剂毒性较大,因此,限制了该方法的应用[Sibi M.P.,Marvin M.,Sharma R.J.Org.Chem.,1995,56,5016–5023]。偶联法是在金属或金属盐的作用下,两分子的单羰基化合物自身偶联生成α-二酮化合物。该反应一般经过自由基反应过程,缺点是该方法一般只能制备对称的α-二酮化合物[Collin,J.,Namy,J.L.,Dallemer,F.,et al.J.Org.Chem.,1991,56,3118–3122]。氧化法合成α-二酮是目前研究和应用最多的一种方法,炔烃或烯烃、酮、α-羟基酮、邻二醇等均可以氧化生成α-二酮化合物。但是,目前已报道的合成方法,存在金属氧化剂昂贵、反应温度较高,而且过渡金属催化剂无法回收利用等缺点。
技术实现要素:
本发明提供了一种光催化α-二酮化合物的新颖制备方法,该方法环境友好、条件温和、操作简便,催化剂可回收并且收率较高。
本发明的技术方案:
一种光催化α-二酮化合物的制备方法,以1,2-二苯乙炔衍生物为原料,在无机半导体催化剂和添加剂的作用下,于光照条件下,在有机溶剂中反应12~18小时,得到α-二酮化合物,合成路线如下:
其中,R1,R2选自氢(H)、烷基(alkyl)、卤素(halogens)、三氟甲基(CF3)、甲酰基(COMe)、甲氧基(OMe)和三甲基硅基(SiMe3),R1和R2相同或不同;
所述的1,2-二苯乙炔衍生物与无机半导体催化剂的摩尔比为1:0.1~1:0.5;
所述的1,2-二苯乙炔衍生物与添加剂的摩尔比为1:0.5~1:2;
所述的1,2-二苯乙炔衍生物的摩尔浓度为0.01mmol/mL~2mmol/mL。
有机溶剂包括四氢呋喃、乙醇、二氯甲烷、丙酮、乙腈、1,4-二氧六环、N,N-二甲基甲酰胺等,优选乙腈。
无机半导体催化剂包括二氧化钛(TiO2)、硫化镉(CdS)、氧化锌(ZnO)、碳化氮(C3N4)、钨酸铋(Bi2WO6)、钒酸铋(BiVO4)。
添加剂包括苯硫酚、甲基苯硫酚、氯代苯硫酚、氟代苯硫酚、甲氧基苯硫酚、硝基苯硫酚以及对应硫醚类化合物。
光源包括5W-12W的蓝光LED,5W-25W的家用白色节能灯以及50W-500W的氙灯。
分离方法包括重结晶、柱层析等。重结晶方法使用的溶剂包括苯、乙醇、石油醚、乙腈、四氢呋喃、氯仿、正己烷、丙酮、乙酸乙酯、二氯甲烷;用柱层析方法进行产物分离时,可以使用硅胶或氧化铝作为固定相,展开剂一般为极性与非极性的的混合溶剂,如乙酸乙酯-石油醚、乙酸乙酯-正己烷、二氯甲烷-石油醚、甲醇-石油醚。
本发明的有益效果是操作简便、条件温和、环境友好、有实现工业化的可能性,并且以较高收率得到α-二酮化合物;利用该方法所合成的α-二酮化合物可以进一步官能化得到各类化合物,应用于天然产物、功能材料及精细化学品的开发与研究。
附图说明
图1是实施例1中1-(4-甲氧基苯基)-2-苯基乙二酮的1H核磁谱图。
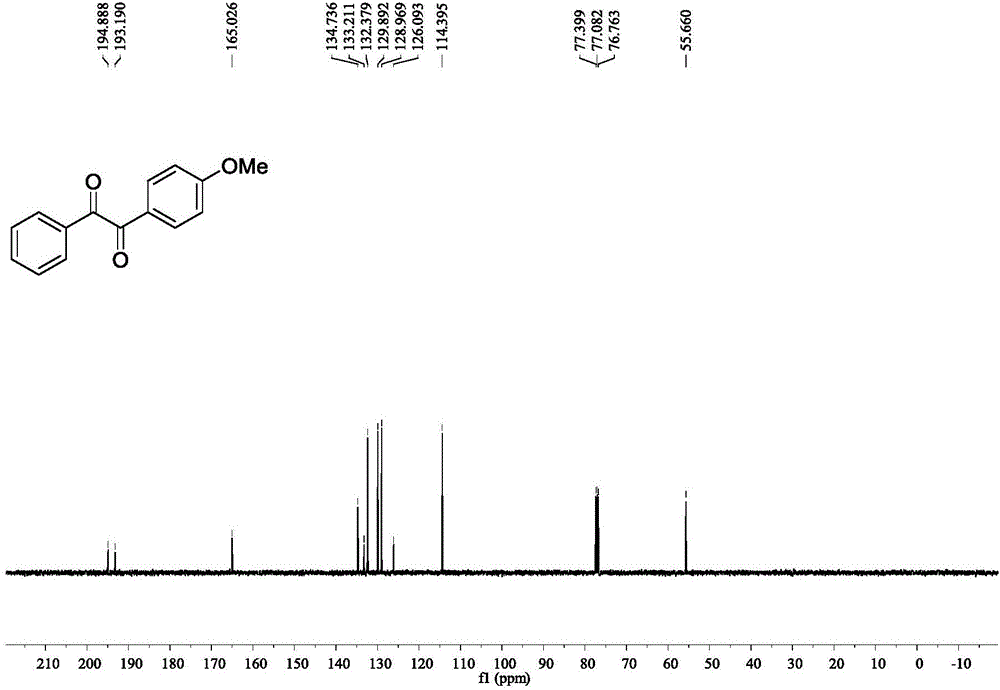
图2是实施例1中1-(4-甲氧基苯基)-2-苯基乙二酮的13C核磁谱图。
图3是实施例2中1-(3-溴苯基)-2-苯基乙二酮的1H核磁谱图。
图4是实施例2中1-(3-溴苯基)-2-苯基乙二酮的13C核磁谱图。
图5是实施例3中1-(3-甲基苯基)-2-苯基乙二酮的1H核磁谱图。
图6是实施例3中1-(3-甲基苯基)-2-苯基乙二酮的13C核磁谱图。
图7是实施例4中1-(4-氯苯基)-2-苯基乙二酮的1H核磁谱图。
图8是实施例4中1-(4-氯苯基)-2-苯基乙二酮的13C核磁谱图。
图9是实施例5中1-(4-乙酰苯基)-2-苯基乙二酮的1H核磁谱图。
图10是实施例5中1-(4-乙酰苯基)-2-苯基乙二酮的13C核磁谱图。
图11是实施例6中1-(4-三甲基硅基苯基)-2-苯基乙二酮的1H核磁谱图。
图12是实施例6中1-(4-三甲基硅基苯基)-2-苯基乙二酮的13C核磁谱图。
图13是实施例7中1,2-二(3-氯苯基)乙二酮的1H核磁谱图。
图14是实施例7中1,2-二(3-氯苯基)乙二酮的13C核磁谱图。
图15是实施例8中1-(1-萘基)-2-苯基乙二酮的1H核磁谱图。
图16是实施例8中1-(1-萘基)-2-苯基乙二酮的13C核磁谱图。
具体实施方式
本发明所述的α-二酮化合物的制备方法,具有原料价格低廉、反应步骤少、反应条件温和、环境友好、便于操作、反应收率高和催化剂可回收利用等优点。
下面结合具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。在本领域内的技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案之内。
实施例1:1-(4-甲氧基苯基)-2-苯基乙二酮的合成
在25mL反应器中,加入1-甲氧基-4-(苯基乙炔基)苯(0.052g,0.025mmol),对甲基苯硫酚(0.031g,0.25mmol),TiO2(0.010g,0.125mmol),乙醇2mL,12W蓝光LED照射下搅拌16h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=50:1)得到1-(4-甲氧基苯基)-2-苯基乙二酮0.041g,产率68%。
1-(4-甲氧基苯基)-2-苯基乙二酮
黄色液体,1H NMR(CDCl3,400MHz)δ7.98–7.93(m,4H),7.65–7.61(m,1H),7.51–7.47(m,2H),7.98–7.93(m,2H),6.98–6.96(m,2H),3.87(s,3H);13C NMR(CDCl3,100MHz):δ194.9,193.2,165.0 134.7,133.2,132.4,129.9,129.0,126.1,114.4,55.7.
实施例2:1-(3-溴苯基)-2-苯基乙二酮的合成
操作同实施例1,由1-溴-3-(苯基乙炔基)苯反应得到1-(3-溴苯基)-2-苯基乙二酮0.056g,产率78%。
1-(3-溴苯基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ8.13(s,1H),7.97(d,J=7.2,2H),7.884(d,J=7.6,1H),7.78(d,J=8.0,1H),7.69–7.66(m,1H),7.55–7.51(m,2H),7.32-7.43(m,1H);13C NMR(100MHz,CDCl3)δ193.6,192.9,137.7,135.2,134.7,132.7,130.6,130.0,129.1,128.6,123.3.
实施例3:1-(3-甲基苯基)-2-苯基乙二酮的合成
在25mL反应器中,加入1-甲基-3-(苯基乙炔基)苯(0.048g,0.025mmol),邻氯苯硫酚(0.036g,0.25mmol),BiVO4(0.040g,0.125mmol),乙醇2mL,12W蓝光LED照射下搅拌16h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=50:1)得到1-(3-甲基苯基)-2-苯基乙二酮0.040g,产率65%。
1-(3-甲基苯基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ7.97(d,J=7.6Hz,2H),7.78–7.76(m,2H),7.67–7.64(m,1H),7.53–7.38(m,4H),2.41(s,3H);13C NMR(100MHz,CDCl3)δ194.8,139.0,135.8,134.8,133.1,130.0,129.0,128.9,127.2.
实施例4:1-(4-氯苯基)-2-苯基乙二酮的合成
在25mL反应器中,加入1-氯-4-(苯基乙炔基)苯(0.052g,0.025mmol),对氯苯硫酚(0.036g,0.25mmol),CdS(0.018g,0.125mmol),四氢呋喃2mL,12W蓝光LED照射下搅拌10h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=50:1)得到1-(4-氯苯基)-2-苯基乙二酮0.046g,产率75%。
1-(4-氯苯基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ7.98–7.92(m,4H),7.67–7.65(m,1H),7.54–7.48(m,4H);13C NMR(100MHz,CDCl3)δ193.9,193.0,141.6,135.1,132.8,131.4,131.2,130.0,129.4,129.1.
实施例5:1-(4-乙酰苯基)-2-苯基乙二酮的合成
在25mL反应器中,加入4-(苯基乙炔基)苯乙酮(0.055g,0.025mmol),对氯苯硫酚(0.036g,0.25mmol),ZnO(0.010g,0.125mmol),四氢呋喃2mL,12W蓝光LED照射下搅拌10h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=50:1)得到1-(4-乙酰苯基)-2-苯基乙二酮0.041g,产率65%。
1-(4-乙酰苯基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ8.07(s,4H),7.98(d,J=7.6Hz,2H),7.71–7.67(m,1H),7.55–7.52(m,2H),2.65(s,3H);13C NMR(100MHz,CDCl3)δ197.2,193.8,193.6,141.3,136.0,135.2,132.7,130.1,130.0,129.1,128.7,26.9.
实施例6:1-(4-三甲基硅基苯基)-2-苯基乙二酮的合成
在25mL反应器中,加入4-(三甲硅基)二苯乙炔(0.062g,0.025mmol),对氯苯硫酚(0.036g,0.25mmol),ZnO(0.010g,0.125mmol),二氯甲烷2mL,20W家用白色节能灯照射下搅拌18h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=100:1)得到1-(4-三甲硅基苯基)-2-苯基乙二酮0.051g,产率72%。
1-(4-三甲硅基苯基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ7.98–7.91(m,4H),7.68–7.63(m,3H),7.52–7.48(m,2H),0.29(s,9H);13C NMR(100MHz,CDCl3)δ194.9,194.7,150.0,134.9,133.9,133.1,133.0,129.9,128.7,-1.42.
实施例7:1,2-二(3-氯苯基)乙二酮的合成
操作同实施例6,由1,2-二(3-氯苯基)乙炔反应得到1,2-二(3-氯苯基)乙二酮0.055g,产率78%。
1,2-二(3-氯苯基)乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ7.96(s,2H),7.83(d,J=8.0Hz,2H),7.65–1.63(m,1H);13C NMR(100MHz,CDCl3)δ192.0,135.5,135.0,134.1,130.4,129.6,128.2.
实施例8:1-(1-萘基)-2-苯基乙二酮的合成
在25mL反应器中,加入1-(1-萘基)-2-苯基乙炔(0.057g,0.025mmol),对氟苯硫酚(0.032g,0.25mmol),BiVO4(0.040g,0.125mmol),二氯甲烷2mL,100W氙灯照射下搅拌18h后,柱层析分离(硅胶,200-300目;石油醚:乙酸乙酯=20:1)得到1-(1-萘基)-2-苯基乙二酮0.020g,产率31%。
1-(1-萘基)-2-苯基乙二酮
黄色固体;1H NMR(CDCl3,400MHz)δ9.30(d,J=8.8Hz,1H),8.12(d,J=8.4Hz,1H),8.04–8.02(m,2H),7.95-7.90(m,2H),7.77–7.73(m,1H),7.68–7.61(m,2H),7.54–7.47(m,3H);13C NMR(100MHz,CDCl3)δ197.2,194.6,136.0,135.1, 134.8,134.1,133.4,129.5,128.6,126.0,124.4。
- 还没有人留言评论。精彩留言会获得点赞!