一种基于比率的生物标记物对及其选择方法与流程
本发明属于生物检测方法领域,具体涉及一种一种基于比率的生物标记物对及其选择方法,更具体的涉及一种选择血浆中ncrnas对的方法,尤其涉及一种能够区分健康对照和肺腺癌的ncrnas对及其具体选择方法。
背景技术:
mirnas是内生的,小非编码rnas,通常18-25个核苷酸长度。它们被发现在mrna转录后调节起着至关重要的作用。mirnas在细胞分化、增殖、凋亡中发挥关键作用,并且参与许多类型的疾病包括癌症,糖尿病,心脑血管和神经疾病。除了mirnas,还有一些其它小非编码rnas,在调节基因表达的很多层面中发挥重要作用,如染色质结构、转录、mrna稳定及翻译,ncrnas包括小snornas,piwi-相互作用rnas(pirnas),短干扰rnas(sirnas)及trnas,这些ncrnas在癌症和其他疾病中被扰乱。例如,snornas包含一个高度丰富的小ncrnas组,和一个在基因剪接和沉默中有ncrna-相似功能的数量有限的snornas组。近期有报道指出3个snornas在非小细胞肺癌病人中显示差异表达,在肿瘤形成中最近的研究表明,三个snornas显示改变表达式在非小细胞肺癌(nsclc)患者,和snora42在肺肿瘤发生中可能作为癌基因。
近年来,一系列研究表明mirnas在如血清,血浆,唾液,乳液,痰液和尿液这些体液中能被检测到,循环mirnas被探测到被外来物质或微泡包裹,或者与特异蛋白如ago-2绑定。一旦在细胞外空间,mirnas能被其他细胞(细胞间通信)占据,由rnases退化,或排泄。尽管mirnas的分泌和整合机制并没有被完全认识,循环mirnas可能参与生理和病理活动。
这些发现为循环ncrnas作为不同种类疾病的非侵入性诊断和预测生物标志物打开了一扇门。由于灵敏度高,特异性强和模板要求量少,目前大多数研究使用测量循环mirnasis逆转录定量pcr(rt-qpcr)方法。因为体液中循环rnas浓度非常低,准确地测量循环mirna的表达是一个巨大挑战。此外,与基因表达相似,系统因素如原始材料数量的变化,样品收集,rna提取,反转录,pcr,这些都会影响最终结果并且引起偏差和定量误差。所以当前,标准化参考对照分子被用于标准化循环mirnapcr数据,目的是公正的评估循环mirna的表达。目前参考对照分子包括内源和外源对照。许多研究者选择使用激增合成rna序列(像c.线虫mir-39和mir-54,或植物mirnas)作为极值参考对照,用于标准化循环mirnaqpcr分析。一系列的内部对照被采用。比如,小核仁rnas(snornas)成员之一,如rnu6b是最早被用于循环mirna数据标准化,但后来被取消了,由于特殊疾病和肿瘤预后。许多研究考虑mirna参照,像mir-16,它在癌症病人血浆样本中显示差异。因为缺乏浓度归一化方法,这会影响不同研究的数据一致性和再现性。因此,当务之急是寻找循环mirna数据的最佳标准化方法。
血浆/血清ncrnart-qpcr实验数据标准化是一个挑战。拿mirna举例,因为从小体积血浆或血清样本(i.e.,100或200μl)中获得的总rna的产量低于分光光度法准确定量的极限值,样品收集,贮存和处理中的误差也影响循环mirna定量分析的精确度和可靠性。鉴于内源或外源参考对照分子中的杂质,当前的实验推荐在rna恢复程序中调整技术差异。对于循环mirnaqpcr分析的标准化,很多研究人员选择在样本中加入合成rna序列(如c.线虫mir-39和mir-54,或植物mirnas)。我们在前期研究中,选择c.线虫cel-mir-54作为一个外部控制,然而发现在测序和rt-qpcr数据中,它不是一个很好的参照。原因在于这些合成的mirnas直接加到血浆中很快被降解,且它加入血浆中后比的内源mirnas稳定性差,因为它们不受内源rnase活性的保护。而且,循环mirnas相对稳定,因为它们受内源rnase活性的保护,也因为它们要么结合于蛋白或包含在核内体之中。
一些研究者为寻求合适的内源参照mirnas(ecm)做出了努力;不管怎样,对于血液mirna定量分析,目前尚未鉴定出足够理想的ecms.比如,mir-16经常被用作参照,但是血清中mir-16的高水平与乳腺癌病人的骨转移相关,并且报道表明内源的mir-16是很差的标准化因子。自chenx等报道let-7d/g/i对于循环mirna数据的标准化是一个很好的内源参照,在实验中我们采用let-7d/g/i进行了测量。我们发现它们在我们的样本中不能稳定表达。chen的样本来自中国人,尽管肺癌样本包含在其中,这可能是我们没有得到相似结果的一个原因。广泛使用的内源参照has-mir-19130在我们的实验中也不是一个很好的参照。我们可以不断的测试更多的内源对照如u637,rnu4438,rnu4839,mir-1640,mir-10330,和mir-23a41,它们是当下被普遍使用的。然而,chen的研究已经发现这些参照得出的结果比let-7d/g/i还差。众所周知,理想的内源参考对照至少满足的条件是:它们在所有样本和实验条件中能稳定表达。这很难证明哪个候选内源分子满足这个条件。
使用比率作为分子标记已经被用于一些疾病中。然而,还没有特别的方法研究提到基于比率的方法应用于循环ncrna测序和rt-qpcr数据标准化。目前,关于循环ncrnas(mirnas)的论文大约99%仍然使用外源或内源参考对照分子来进行循环pcr数据的标准化。一些研究仍在极度寻求循环mirna数据标准化的更好参考对照。
技术实现要素:
近期有研究已经表明循环ncrna,如mirnas,是稳定的且可被作为分子标记用于人类疾病诊断与预后。然而,由于血液中的循环ncrnas浓度很低,使用新一代测序和定量实时rt-pcr进行血浆/血清ncrna实验中,数据标准化是一个挑战。目前标准化方法基于合成外源性标准对照或寻找内源性mirna对照是不合适的,因为他们没有稳定表达,从而没有找到可靠的差异表达显著的ncrnas。
针对上述现有技术的缺陷,本发明提供了一种基于比率的标准化方法,用于代替以单个ncrnas作为生物标志物,对相同的样本我们计算任意两个ncrnas的比率,并使用产生的比率作为生物标志。
一方面,本发明提供一种选择生物标记物对的方法,包括以下步骤
(1)确定生物样品中ncrnas的种类;
(2)确定生物样品中ncrnas的含量;
(3)计算每个生物样品中任意两种ncrna之间的比率;
(4)根据多个生物样品组中每种ncrna的组平均值,计算任意两种ncrna组平均值之间的比率;
(5)采用支持向量机循环特性选择(svm-rfe)算法选择最佳ncrnas对;
(6)以ncrnas对的比率作为将样品分组的指标。
本发明所述的选择生物标记物对的方法,其中所述生物样品为血浆;所述生物样品组至少包括正常样品组、疾病样品组,优选所述疾病样品组包括癌症样品组、良性肿瘤样品组;所述ncrna包括mirna、snorna、pirna、sirna及trna。
本发明所述的选择生物标记物对的方法,其中所述步骤(1)包括rna提取和小分子rna测序。
本发明所述的选择生物标记物对的方法,其中所述rna提取是将血浆用trizol试剂提取,加入二氧化硅膜封闭柱内吸附,清洗后收集吸附的rna。
本发明所述的选择生物标记物对的方法,其中通过smartersmrna-seq法进行测序,具体包括将rna样本进行3’接头连接、5’rt引物退火、5’接头连接、反转录、pcr扩增。
本发明所述的选择生物标记物对的方法,其中所述步骤(2)中通过反转录和pcr定量(rt-qpcr)确定血浆ncrnas的含量,优选使用taqmanmirna试剂盒进行qrt-pcr检测。
本发明所述的选择生物标记物对的方法,其中所述步骤(3)根据rt-qpcr的数据,同一样品中2个小分子ncrna含量的比率(ncrna1/ncrna2)采用比较ct法(2-δct)进行计算,δct=ctncrna1-ctncrna2。
本发明所述的选择生物标记物对的方法,其中所述步骤(4)包括将血浆ncrna浓度通过log2转换,采用spss20.0软件进行非配对t检验,比较不同生物样品组之间平均ncrna比率,显著p-值设置为0.05。
本发明所述的选择生物标记物对的方法,其中所述步骤(5)中支持向量机循环特性选择(svm-rfe)算法包括:
a、初始化数据集包含的特征;
b、训练数据集的svm;
c、根据ci=(wi)2排名特征;
d、剔除低等级特征的50%;
e、返回步骤b。
本发明所述的选择生物标记物对的方法,其特征在于:所述的ncrna还可以用其他生物标记物替换,所述其他生物标记物包括mrna、dna、蛋白质、代谢产物。
第二方面,本发明提供一种由所述的选择生物标记物对的方法选择获得的生物标记物对。
本发明所述生物标记物对其选自以下的组mir378a-3p/mir126-5p、sno-dr119/trna-thr-acg、sno-aca33/mir378a-3p、trna-thr-acg/sno-u57、trna-thr-acg/mir378a-3p。
第三方面,本发明提供所述生物标记物对在制备肺腺癌诊断试剂中的用途。
值得注意的是,本发明所述选择生物标记物对的方法是以离体的生物样本为实施对象,但其直接目的是为了选出生物标记物对(以便于后续的研究或应用),而并非对生物样品来源的个体进行诊断或治疗,因为选择生物标记物对的方法中所使用的生物样本其来源个体的健康状况(如肺腺癌患病情况)是已经通过其他方法诊断确定了的。因此本发明所述选择生物标记物对的方法本身就是非临床诊断目的的方法。
虽然根据本发明的原理,本发明选出的生物标记物对能够用于将生物样本材料按来源个体的健康状况进行分类并具有巨大的临床应用价值;但这仅表明本发明所述方法获得的生物标记物对的临床应用可能涉及临床诊断方法,而选择生物标记物对的方法本身,则不属于任何临床诊断方法。
与现有技术相比,本发明取得了以下有益的技术效果:
(1)本发明提出的基于比率的标准化循环ncrna数据的方法,采用ncrnas的比率作为分类依据,相比于采用单一ncrna,ncrnas的比率数量更多、差异更显著、能更准确的反应真实值。例如对相同的样本,我们首先计算任意两个ncrnas的比率,然后比较不同组之间的比率表达水平而不是单个ncrna水平进行比较。因为2个ncrnas采用相同的样本,在同等条件下同时表达,2个ncrnas比值的相对表达水平将反映比较的真实值。
(2)本发明在数学逻辑上证明了本发明所述方法是正确的,它独立于任何外部或内部参考控制分子,并优于任何现有基于外部或内部控制标准化方法。这一比率策略将在临床应用循环ncrna作为人类疾病的生物标记方面提供一个实际的方法。本发明在数学上证明基于比率的方法优于任何基于内源或外源对照的标准化因子的方法。基于内源或外源对照的标准化方法有两个假设。首先,假设在相同样本中的待测mirna和内部控制受到相同的系统因子影响;第二,假设不同样本中真实内控值一样。而基于比率的方法仅仅假设相同样本中的不同mirnas有相同的系统因子,因此,从数学上清楚地证明了基于比率的标准化方法优于基于参考对照的标准化方法,因为很难了解第二个假设真实与否。
(3)基于比率的生物标记引物对增加了寻找有临床意义生物标记的几率。基于比率的标准化方法能够在不同疾病组中发现更重要的不同ncrna候选标记。这在逻辑上也易于理解,比如,在健康正常组和癌症组中给定mirna1/mirna2的比值,如果在癌症组对比正常组中有一个上调的倍数改变,而mirna2在癌症组对比正常组中有一个下调的倍数改变。这样的话癌症组和正常组之间的mirna1/mirna2倍数改变比单独mirna1或mirna2更大。因此当我们不能找到有重大变化的单一标记时,基于比率的方法将增加我们寻找有临床意义生物标记的机率。
(4)本发明运用所述方法找到了5个循环ncrna比率,这5个ncrna组合有100%预测准确性来区分肺腺癌与正常人,并且本发明不仅测试mirnas,也测试其它类型的ncrnas,如snornas和trnas。
附图说明
图1:外源性对照cel-mir-54的阅读数。
7个血浆样本集合(15样本/集合)被用于小分子rna的测序,在rna提取和测序前,合成的外源性cel-mir-54等量添加到7个血浆样本集合中。lc代表正常健康对照(2个样本集),be代表良性(2个样本集),ad代表肺腺癌(2个样本集)并且sc代表鳞状细胞肺癌(1个样本集)。
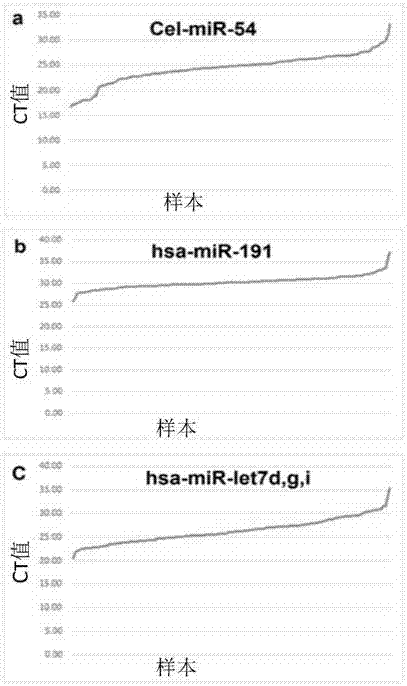
图2:癌症和非癌症样本中外源和内源性参考对照的rt-qpcrct值。ct值的分类是基于总数为129的血浆样本量,这些样本包括肺癌,良性和正常健康对照。(a)129例样本中外源c.线虫cel-mir-54ct值。(b)129例样本中内源参考对照has-mir-191ct值。(c)129例样本中内源参考对照平均has-let-mir-let7d,g,ict值。
图3:存在差异的单个ncrna数量和ncrna比值的数量。
x轴代表所有的可测量特性(mirna比率或mirna),正常健康对照vs肺腺癌中可区分数量,正常健康对照vs良性及良性vs肺腺癌。非配对t-test被用于鉴定可区分mirna或mirna比率。pvalue<=0.05且倍数改变切断为2.0。
图4:正常样本和肺腺癌样本中代表性ncrna比率的表达值。
adenocarcinoma:肺腺癌;normal:正常。血浆中每一个个体ncrna采用实时定量rt-pcr被测量,相同样本中2个ncrnas的比率被计算成(2-δct),其中δct=ctncrna1–ctncrna2,所以-δct=log2(ncrna1/ncrna2)。(a)mir378a-3p/mir126-5p.(b)sno-dr119/trna-thr-acg.(c)trna-thr-acg/sno-u57.(d)trna-thr-acg/mir378a-3p.(***p<0.001)。
图5:肺腺癌与正常对照组血浆样本通过5个配对ncrna比率标记的区分。
基于5个配对标记的双向分级群聚被执行来展现群体聚类。50例肺腺癌样本(adeno)和29例正常健康对照(normal)被用来进行实时rt-qpcr。彩条表示标记的表达值。
具体实施方式
以下结合具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
实施例1
样本组和血浆样本收集
在北京大学人民医院,在肺癌生物库中我们收集大约1250名肺癌患者标本,从这些标本中我们选择了一个包括130名患者的小组用于此次研究,这130例标本例包括50例早期阶段(阶段i,ii)肺腺癌,和15例鳞状细胞肺癌(scc),35例良性,30例正常。早期腺癌和鳞状细胞肺癌scc样本入选标准包括:仅患未有远端转移的胸病;我们开始采血前1年内未有术前化疗或放疗;至少2年的临床跟踪数据。良性样本候选病人入选需经低剂量计算机扫描显示(ldct)患一系列非肿瘤的肺小病(如息肉,错构瘤,和炎性病变)。所有入选良性病人和正常人接下来每年都进行ldct,并且至少2年内不患癌。这些病人和对照的人口统计信息如补充表1。癌症,良性和正常样本在年龄,种族,性别和吸烟状况方面尽可能的匹配。正常组也被称为高风险组,这组人在随机取样前有30年以上吸烟史并且戒烟史低于15年。所有病人数据都获得书面正式同意,并且符合北京大学人民医院伦理审查委员会的相关要求。
所有血浆样本用edta抗凝管收集,4000rpm离心10min,接着12,000rpm高速离心15min完全去除细胞碎片。上层血浆在分析前贮存在-80℃。所有样本在首次诊断时收集。
实施例2
rna提取和测序
rna提取如前所述。血浆总rna,包括血浆小分子rnas的提取采用小份mirneasykit(qiagen,valenciz,ca)。简言之,0.5ml血浆用无rnase的水1:1稀释(总共1ml),完全分层后每1ml样本体积加入3ml
在此研究中,我们采用加利福尼亚希望市的一种下一代测序来研究血浆样本,这种技术如前所述。简言之,为节约成本和样本,我们首先分析小分子rna测序(smrna-seq),从而鉴定micrornas和其它一些循环小分子非编码rnas(sncrnas)。我们选用7个样本集,这7个样本集中包括30例高风险健康对照(正常),30例良性结节病灶(良性),30例肺腺癌,和15例鳞状细胞肺癌。正常,良性和癌症样本在年龄,性别,种族和吸烟状况方面都是匹配的。训练集样本(来自北京大学人民医院,但很不幸,我在进行pcr时丢失了一例正常样本)按预期收集。验证集样本按预期收集。除鳞状细胞癌组,每组两个样本集(每个样本集15例)都进行smrna测序,每个样本集中同等混匀500μl血浆。每个样本2000万读取与约90%的读取与人类基因组比对。
文库构建预备实验,6μl血清rna提取洗脱液,预备实验按照指示草案进行了少量修改后进行。一个mirna文库构建是由每个rna样本通过3′接头连接,5′rt引物退火,5′接头连接,反转录,和pcr扩增。12份样本文库等量混合,聚合后用cbot(illumina)读取每个通道的单一读取流动细胞的浓度为10.5pmol。hiseq2500(illumina)测序设置为50个循环。多路分解原始测序数据并使用casavav.1.8.2生成fastq文件。
从fastq文件,通过将接头与测序阅读框局部比对,3'测序接头将被移除。我们采用切除接头软件来移除3'接头。接头移除后,所有序列都有少于15bps长度的缺失。每个文库的读取将总结成量化fasta格式的标签。因为bowtie,fasta阅读将被映射到基因组。为消除模糊映射记录,只有最少的比对不匹配的独特映射位点将会被记述,最多允许两个不匹配。不同文库的表达谱将将取决于人类ncrnas映射清洁读回。对于每一个映射轨迹注释来自多个ncrna数据库。
实施例3
反转录和实时pcr
使用taqmanmirna实验试剂盒(appliedbiosystems,美国),根据制造商提供的草案,ncrnas将被测量。简言之,大约30ng丰度的rna使用taqmanncrna反转录试剂盒(appliedbiosystems,美国)被反转录,反应体积为15μl。ncrnas的表达水平被一式三份进行qrt-pcr定量分析,使用taqmanmicrorna实验试剂盒(appliedbiosystems,美国),eppendorfiplex4系统(eppendorfnorthamerica,hauppauge,ny)。为绕过标准化问题,我们采用相同比率策略来替代标准化,从而减少实验误差。
实施例4
统计分析
对于相同样本,我们计算任意两个ncrnas的测序和rt-qpcr数据比率。对于rt-qpcr数据,2个小分子ncrna(ncrna1/ncrna2)比率的表达水平的计算采用比较ct法(2-δct),对相同的样本,δct=ctncrna1–ctncrna2。血浆ncrna浓度被log2转换后,我们采用spss20.0软件进行非配对t-检验,比较腺癌组,良性病人和正常对照组之间平均ncrna比率,显著p-值设置为0.05。用spss20.0软件进行卡方检验,比较训练和验证阶段样本性别,种族和癌症分期的分布,年龄用t-检验,显著p-值设置为0.05。采用支持向量机循环特性选择(svm-rfe)算法来选择最佳ncrnas。svm-rfe48是为特定学习目标选择特征子集的一种算法。它的基本算法是:(1)初始化数据集包含的特征,(2)训练数据集的svm,(3)根据ci=(wi)2排名特征,(4)剔除低等级特征的50%,(5)返回步骤2。在每个rfe步骤4,一些特征因svm分类模型的主动变量被舍弃。特征根据相关标准被消除,这一标准与它们支持判别函数相关,在每一步svm被再训练。根据特征选择算法选择的ncrna比率被用于使用支持向量机(svms)的分类。5倍十字交叉验证过程包括内部和外部验证。我们使用了预测性能指标,包括敏感性,特异性,阳性预测值(ppv),阴性预测值(npv)和roc曲线下面积(auc)来判断预测精度。
实施例5
基于比率的对循环ncrna剖面数据标准化的方法独立于任何内部或外部标准对照
因为外源和内源对照对循环ncrna剖面数据标准化都不可信(图2),我们测试了一个基于比率的,用于循环ncrna剖面数据标准化的方法。首先,我们计算相同样本中任意两个ncrnas的比率,接着比较不同组别中表达水平比率而不是比较单一ncrna的表达水平。拿mirna和内控(ic)举个例(表1)。
表1基于归一化方法的率
*正值代表在癌症中上调,负值代表在癌症中下调。
mirna1的表达值在正常样本和癌症样本中分别是4和8,两组间倍数变化为2(行1);内部对照1(ic1)的表达值在正常样本和癌症样本中分别是2和4(行2)。如果mirna1通过ic1标准化,正常样本和癌症样本之间的倍数改变是1(行5);如果mirna1通过内控2(ic2)标准化,正常样本和癌症样本之间的倍数改变是-4(平均下调4倍)。可以看到,没有标准(行1)或使用不同的内控(ic1或ic2),正常样本和癌症样本之间的倍数改变不一样。与mirna2相似,我们得到不同的倍数改变结果(如行2,6和8)。如果我们首先通过ic1来标准化mirna1和mirna2,然后计算ic1标准化mirna1和mirna2的值的比率,正常样本的值为0.5,而癌症样本的值为2,倍数改变为4(行9)。有趣的是,如果我们通过ic2来标准化mirna2(行10)或不用任何对照(行11),然后计算相同样本同两个mirnas的比率,正常样本的比率值仍然为0.5(行10和11),而癌症样本的值也为2(行10和11),倍数改变仍为4(行10和11)。结果显示,不管我们用哪种内控方法,相同样本中任意两个mirnas的比率不变。所以,对于mirna剖面数据的标准化,我们只能计算相同样本中任意两个mirnas的比率(行11),这是完全独立于任何内源或外源对照的。
实施例6
基于比率的数据标准化方法在数学逻辑上是正确的
从表1,我们已经了解到,基于比率的标准化方法是有意义的。在此,我期望从数学逻辑上证明该法是正确的。此外,我们采用mirna作为一个例子,我们的最终目标是试图发现生物学上真实mirna值(turemirna),然而,通常我们从实验中得到的观察mirna(obsmirna)值不是真值。事实上,obsmirna值是truemirna被嵌入不同的系统因子得到的结果。rt-qpcr实验中,系统因子可能包括rna提取(i),反转录(r),pcr(p),不同时间(t)等等。因此,在一个具体案例中如s1,设置如下:
(1)obsmirna1=truemirna1*is1*rs1*ps1*ts1
同理,我们假设在相同样本中对于mirna2的系统因子一样,在同一s1中的obsmirna2同样设置如下:
(2)obsmirna2=truemirna2*is1*rs1*ps1*ts1
因此,(3)obsmirna1/obsmirna2=truemirna1/truemirna2
从(3)行,我们可以清楚看到,在相同样本中,2个观察mirnas值的比率等于2个真实mirnas值的比率。因此,我们从数学上证明,在相同样本中,2个观察mirnas比值能够真实反映2个mirnas生物学价值,这个生物学价值是我们期望测量的。
因此,pcr值是ct值,ct值实际上是对数值。从公式(4),我们了解到,2个mirnas的对数比值实际上是2个mirnas的2个ct值的差值,这使得计算更简单并且使得临床上使用这些基于rt-qpcr的数据更方便。(4)log2(obsmirna1/obsmirna2)=log2(2-ctmirna1/2-ctmirna2)=log2(2-ctmirna1/2-ctmirna2)=log2(2-ctmirna1+ctmirna2)=ctmirna2-ctmirna1
实施例7
数学上,基于比率的标准化方法优于内源或外源对照标准化方法
虽然,我们从数学角度证明基于比率的标准化方法在数学逻辑上是证确的,人们可能有疑问,因为我们假设在相同样本中,对不同的mirnas系统因子是一样的。在理论上它是对的,因为那两个mirnas是在相同样本中,应该嵌入相同的系统因子。实际上,基于标准化方法的参考对照也做了同样设置。
从数学角度进一步分析比较基于比率的标准化方法与内源或外源对照标准化方法:
(1)obsmirna1s1=truemirna1s1*is1*rs1*ps1*ts1
可如下设置
(2)is1*rs1*ps1*ts=factor1
然后,样本1(s1)中的mirna1真实值为
(3)truemirna1s1=obsmirna1s1/factor1
同样地,样本2(s2)中的mirna1真实值为
(4)truemirna1s2=obsmirna1s2/factor2
同样地,样本1(s1)和样本2中的内控(ic)真实值为
(5)trueics1=obsics1/factor1
(6)trueics2=obsics2/factor2
因此基于(5)和(6),我们得到
(7)factor1=obsics1/trueics1
(8)factor2=obsics2/trueics2
用(7)中factor1代替(3)中factor1,(8)中factor2代替(4)中factor2,我们得到
(9)truemirna1s1=(obsmirna1s1/obsics1)*trueics1
(10)truemirna1s2=(obsmirna1s2/obsics2)*trueics2
假设(11)trueics1=trueics2
因此
(12)truemirna1s1=obsmirna1s1/obsics1
(13)truemirna1s2=obsmirna1s2/obsics2
(12)和(13)中公式是目前基于内源性或外源性对照的标准化方法。它考虑到在相同样本中,通过内部参照(ic)得到的一个观察mirna的归一化值是mirna的真实值。为得到这个值,这里有两个假设:第一,假设在相同样本中,待测mirna和内控受相同系统因素的影响(如(2)和(5)或(4)和(6)),第二,假设在不同样本中,真实内控值是一样的(如(11))。然而,这很难了解到第二个假设是对的或错的。基于比值的方法仅仅假设在相同样本中不同mirnas有相同的系统因子,因此,我从数学角度很清楚地证明了,基于比率的标准化方法优于基于参考对照的标准化方法。
实施例8
基于比率的标准化方法能在不同疾病组中找到更多重要的可作为候选标记的差异ncrna
最初我们假设,对于循环rt-qpcr数据采用基于比率的标准化方法,因为加入外源性对照对测序数据的标准化失败了。拿mirna作为一个例子,一个mirna至少20次阅读,在测序样本中我们找到了631个成熟mirnas。接着,我们计算相同样本中任意两个mirnas的比率,我们意外地得到198765个比率(图3),在充分地增加了我们在不同疾病组中寻找候选mirnas的数量,这些候选mirnas是差异表达的配对比值分子。为得到一个差异表达mirna比值表,我们在样本集中进行癌症与正常组,癌症与良性组,良性与正常组差异表达分析。倍数改变>=2且p值<=0.05,我们发现了大量显著改变的成熟的mirna比率(mirna/mirna),其中正常与癌症对比组中有30,989个,正常与良性对比组中有12,701个,良性与癌症对比组中有7,044个。这些显著改变的比率的数量比3个组别中单一mirnas改变的数量要多,单一mirna数据标准化是基于全局中值的(图3)。
实施例9
基于比率的ncrna生物标记用于区分健康对照与肺腺癌
为测试这些基于比率的可以区分肺癌与非肺癌样本的候选ncrnas,从测序数据中,首先我们选择大约20对配对明显的ncrna比率来比较正常组与癌症组,29例正常样本,50例肺腺癌早期样本,这些肺腺癌样本在种族,性别和吸烟状况方面配对。使用支持向量机回归特征消除(svm-rfe)特征选择和svm分类算法,我们发现一个由5个ncrna比率的组合,可使所有测量参数达到预测准确度100%,测量参数包括敏感性,特异性,阳性预测值(ppv),阴性预测值(npv)和roc曲线下面积(auc)。图4显示代表性ncrna比率分子标记在50例肺腺癌和29例正常样本中的表达值。图5描述的是即使使用无监督的分级聚类,肺腺癌与正常样本可以区分且没有一个单一样本的错分。
对比例1
外加c.线虫cel-mir-54对循环小分子rna测序不是好的标准控制
为了确定循环小分子ncrna标记对肺癌的检测,我们执行全部基因组水平的小分子ncrna测序(smrna-seq),使用基于人类血浆样品的样品集来节省成本和样品。我们首先进行smrna-seq来确定血浆中小分子核糖核酸micrornas和其他一些循环小分子非编码rnas(sncrnas),使用的7个样品集包括30例高危健康个体(健康对照),30例良性结节病变,30例早期肺腺癌和15例鳞状细胞肺癌(scc)。每个样本集包含15例样本。正常对照、良性和癌症样本在年龄、性别、种族和吸烟状况方面相匹配。样本在拉什大学医学中心按预期采集。对照组,良性组和腺癌组采用两个样本集,scc用一个样本集。每个样本集包含500μl等量混合血浆,被用于smrna-seq。这一过程在希望市(ca)illumina下一代测序平台完成。大约2000万读取样本生成数据的90%与人类基因组比对。
自从c-线虫cel-mir-5429是人体没有的,在测序中,它可作为一个外部加入对照。rna提取前,每个样本集中添加等量cel-mir54。所有样本集中,我们希望读取到等量cel-mir-54。如图1所示,7个样品集中每个集读取到的cel-mir-54数据大不同。一个肺腺癌组读取到最大数值是200。scc组读取到数值为0。由此,我们认为外加cel-mir-54对照,对smrna-测序数据的标准化不达标。
对比例2
外加c.线虫cel-mir-54作为小分子ncrna循环定量rt-pcr(rt-qpcr)的对照所得数据的标准化也不达标
假设外源c.线虫cel-mir-54是小分子ncrna循环rt-pcr(rt-qpcr)数据标准化的一个外加对照。我们挑选129例样品(29例健康对照,50例肺腺癌,35例良性和15例scc),进行cel-mir-54的rt-qpcr。等量cel-mir-54添加到等量血浆(200μl)中,这些血浆中单个血浆样本是在rna提取前。如图2a所示,我们发现已经发布的外控cel–mir-54的ct值是非常不稳定的;ct值大致范围在14—34。最高和最低ct值相关在约20个ct,与原始数据有40倍的差异。因为添加了等量cel-mir-54,我们期望对同一样本有相似ct值。因此,我们认为外加c.线虫cel-mir-54作为小分子ncrna循环定量rt-pcr(rt-qpcr)的对照所得数据的标准化也不达标。
对比例3
内源性对照对小分子ncrna循环定量rt-pcr(rt-qpcr)数据的标准化不达标
我们用外源加入对照如cel-mir-54对循环ncrnart-qpcr数据的标准化失败了,我们想是否可用内源对照来标准化循环ncrnart-qpcr数据。基于已经发布的报告,我们选择has-mir-19130和has-mirnas,let-7d,let-7g和let-7i31作为我们的内源性对照。我们使用与做外源对照cel-mir-54(图2)时相同的血浆,从相同体积(200μl)血浆样本提取等量体积rna(约2μl),并对相同129份样本,采用内源性对照来进行rt-qpcr。如图2所示,已发布的内源对照包括has-mir-191ct值(图2b)和has-mirnas,let-7d,let-7g和let-7i(图2c)的平均值同样分布不同且不稳定。因此,我们认为选择参考对照作为循环ncrnart-qpcr数据的标准化不合适。
上述说明并非对本发明的限制,本发明也并不限于上述举例。本技术领域的普通技术人员在本发明的实质范围内,作出的变化、改型、添加或替换,也应属于本发明的保护范围,本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!