一种核酸扩增反应引物的设计方法及其应用与流程
本发明属于生物技术领域,涉及一种核酸扩增反应引物的设计方法及其应用,所述方法是一种既可以在特定基因或区域,也可以在全基因组范围内设计出具有通用性和特异性的核酸扩增反应引物的方法。
背景技术:
核酸扩增技术已成为当前分子生物学研究和应用中使用最多、最广泛的手段之一,核酸扩增的目的在于针对目标序列,利用引物组扩增一段不含任何副产物(非特异性DNA)的特异DNA片段,因此引物设计是核酸扩增技术中至关重要的一环。基于核酸扩增技术的目的,扩增反应的引物组首先需要具备特异性,即该引物组不能扩增非目标序列;为了提高效率,有时同时需要引物组具有通用性,即引物组可以应用于多个目标序列(多个目标基因或区域,或多个目标基因组)。
目前常用的引物设计软件一般只能在预先设定的一个基因区域内设计引物,也就是说,用户必须首先寻找到具有特异性和通用性的序列(通常的做法是,从文献中获取物种特异性基因,并根据其保守的范围确定其通用性),然后将其输入引物设计软件如Primer、PrimerExplorer V4等。然而,从文献中获得的所谓“物种特异性基因”往往基于滞后的知识,并未基于不断增长的基因组数据进行必要的更新,导致基于该基因序列获得的引物在实际应用中不一定能确保其特异性和通用性,导致传统方法引物设计的成功率偏低。表1举例展示了现有技术阪崎克罗诺杆菌检测方法中存在的引物特异性不够的问题。也就是说,现有技术方法中所使用的阪崎克罗诺杆菌特异序列实际上并非阪崎克罗诺杆菌所特有,即,有可能将非阪崎克罗诺杆菌错误地认定为阪崎克罗诺杆菌。出现这类问题的原因之一是未利用现有基因组数据库中对设计的引物进行包括诸如特异性检测和判定等的有效性检验。类似的问题同样也存在于通用性的确认。
表1阪崎克罗诺杆菌的现有检测方法中引物的特异性分析
注:a)将专利中引物F3和B3间的序列与阪崎克罗诺杆菌的三个基因组(GI号分别为156932229、449306535和389839000)进行Bowtie比对,确定检测区域在GI号156932229基因组中的位置(星号*表示引物F3和B3间的序列可比对到GI号156932229基因组中的7个重复区域,本表中仅列出1个区域)。专利CN201310287426.5中四套引物的扩增区域无法定位,文本中也没有给出扩增靶基因为何种基因。b)将检测区域序列在公共数据库资源中进行Blast比对,引物区域匹配程度越高,特异性越差。
因此,亟需一种新的方法,不但可以在特定基因或区域,而且可以在全基因组范围内高效率地设计出具有通用性和特异性的引物,满足检测、扩增等对确保具有特异性和通用性的引物的需求。
技术实现要素:
本发明创新提出了一种核酸扩增反应引物的设计方法。该方法既可以针对特定基因或区域的序列,也可以针对全基因组序列进行引物设计。该方法设计得到的引物能够同时通过物理化学性质检测和通用性、特异性检测。本发明方法得到的引物可以扩增多个目标序列(包括含多个目标基因、多个目标区域、或多个目标基因组);同时,得到的引物无法扩增非目标序列。本方法充分利用了公共资源中的基因组学数据,并采纳恰当的数据分析方法和工具,能够在目标序列(包括目标基因、目标区域、或目标基因组)上高效率地进行引物设计,获得具有通用性和特异性的引物,其可靠性超过传统的仅针对特定基因序列的引物设计方法, 本发明确保了所获得的针对该目标序列的引物的特异性和通用性。
本发明提出一种新的核酸扩增反应引物设计方法,该方法可以在特定的目标基因或区域、或者目标全基因组范围内设计出满足通用性和特异性的引物,能够广泛地应用于PCR、实时定量PCR、等温扩增(如LAMP等)、以及其它核酸扩增反应类型所需要引物的设计。
本发明提出的核酸扩增反应引物的设计方法,包括:(1)确定引物的目标序列和非目标序列;(2)针对目标序列设计单条引物;(3)引物序列比对;(4)根据核酸扩增反应的类型及其对引物组合方式的要求,将单条引物组合成引物组;(5)对引物组进行物理化学性质、通用性以及特异性检测和判定;(5)输出符合条件的引物组。
本发明核酸扩增反应引物的设计方法,包括以下步骤:
(1)初始化:将由引物的所有目标序列组成的集合定义为集合G_1,将由引物的所有非目标序列组成的集合定义为集合G_2;
(2)设计单条引物:根据核酸扩增反应的类型,设定描述引物特性的各项参数,以集合G_1中的某一目标序列为引物设计的候选序列,计算得出符合条件的单条引物,并记录每条引物的信息;
(3)引物序列比对:将设计出的单条引物分别与集合G_1和集合G_2进行序列比对,记录比对信息;
(4)单条引物组合:根据核酸扩增反应的类型及其对引物组合方式的要求,将单条引物组合成引物组;
(5)引物的检测:对引物组进行物理化学性质、通用性以及特异性检测和判定;
(6)输出符合条件的引物组,即得到所述核酸扩增反应的引物。
其中,步骤(1)中,所述集合G_1是由引物的所有目标序列组成,所述集合G_2是由引物的所有非目标序列组成。
所述目标序列是指引物设计的目标基因或目标区域、或目标基因组序列。
所述非目标序列是指除目标基因或目标区域之外的序列、或除目标基因组之外的其它基因组序列。
在具体实施方案中,当核酸扩增反应的检测对象为特定某物种时,目标序列 可以是待测物种全基因组,非目标序列则是其它物种或其它相关物种的基因组。
在具体实施方案中,当核酸扩增反应目标为特定基因或区域时,目标序列为待测基因或区域的DNA序列。根据实施方案对特异性的不同要求,非目标序列可以是该目标序列之外的其它任意所有序列,也可以是所在基因组中除目标序列之外的其它序列。
例如,当实验目的为检测混合的微生物基因组中是否存在某种特定微生物X,目标序列是X微生物的各菌株基因组序列,而非目标序列是除X微生物之外的其它微生物基因组序列。例如,当实验目的为设计恰当的引物以扩增出小鼠、大鼠和人的某一基因序列X,则目标序列是小鼠X基因、大鼠X基因和人X基因,非目标序列是小鼠基因组中除X基因之外的其它序列,大鼠基因组中除X基因之外的其它序列,和人基因组中除X基因之外的其它序列。
其中,步骤(2)中,所述根据核酸扩增反应的类型,是指引物的应用范围,可以为聚合酶链式反应(PCR),实时定量PCR,也可以为等温扩增反应,包括环介导等温扩增(Loop-Mediated Isothermal Amplification,LAMP)等,以及其它核酸扩增反应类型。所述“描述引物特性的各项参数”,包含但不限于引物的长度、GC含量、Tm值、3’端稳定性、5’端稳定性和/或二级结构稳定性等物理化学性质,在具体实施方案中根据实际情况确定适当的一项或多项参数的组合。所述“记录每条引物的信息”,是指记录包括引物在所述特定目标序列中的位置、正负链信息(即来源于正链还是负链)和/或引物的长度等信息,在具体实施方案中根据实际情况确定适当的一项或多项参数的组合。
其中,步骤(3)中,所述序列比对,可以使用Bowtie、Bowtie2、BWA、SOAP等短序列快速比对软件,也可以为Blast、Blat等长序列比对软件,优选地,为Bowtie、Bowtie2、BWA、SOAP等快速比对软件。所述“记录比对信息”,是指每条引物在集合G_1和集合G_2中每个序列上面的位置、正负链信息(即来源于正链还是负链)等信息。
其中,步骤(4)中,所述根据核酸扩增反应的类型,是指引物的应用范围,可以为聚合酶链式反应(PCR),实时定量PCR、也可以为等温扩增反应,包括环介导等温扩增(Loop-Mediated Isothermal Amplification,LAMP)等,以及其它核酸扩增反应类型。所述对引物组合方式的要求,是指单条引物在所述特定目 标序列上的位置、正负链信息(即来源于正链还是负链),以及单条引物间的距离等。
其中,步骤(5)中,所述对引物组进行物理化学性质检测,是指检测各条引物在每个序列上的位置关系与正负链信息(即来源于正链还是负链)、引物的GC含量与扩增区域的GC含量的关系、引物的Tm值与扩增区域的Tm值的关系、引物之间是否发生相互作用等。所述通用性检测和判定,是指检测该引物组能否根据检测需求应用在G_1集合中的多个目标序列,若符合检测需求,则通用性检测结果为“通过”;若不符合检测需求,则通用性检测结果为“未通过”。所述特异性检测和判定,是指检测该引物组能否用于集合G_2中的任意一个非目标序列,若不能用于集合G_2中的任意一个非目标序列,则特异性检测结果为“通过”,若能用于集合G_2中的一个或多个非目标序列,则特异性检测结果为“未通过”。若未通过上述物理化学性质、通用性及特异性三项检测的,则放弃该引物组,继续筛选合适的引物组。
其中,步骤(6)中,所述输出符合条件的引物组,是指输出前步骤中获得的能够同时通过物理化学性质检测、通用性和特异性检测的引物组中每一条引物的位置、长度、正负链信息(即来源于正链还是负链)、以及该引物组的通用性情况(即可以用于哪些目标序列上)等信息。系统将符合条件的引物组按照通用性程度排序,通用性越强,排名越靠前,系统按此顺序输出符合条件的引物组。其中,引物组的数量可预先设定。
本发明还提出了按上述方法设计得到的引物,所述引物包括引物组,所述引物组为:
F3:5’-GGATTAGGAAATATACGACGC-3’(SEQ ID NO:1)
FIP:5’-AGCAAAACCCATTGGTACCTTATACTGTCTATGGGCACAA(SEQ ID NO:2)
BIP:5’-TGAATATGATCCCCCGGCTTCTGGGAGGAAATATCGCTAA-3’(SEQ ID NO:3)
B3:5’-AGCATATATGATACGGGGAA-3’(SEQ ID NO:4)。
本发明还提出了按上述方法设计得到的引物,所述引物包括引物组,所述引物组为:
P1:5’-GGAGCCTGCTCAGTCTGTT-3’(SEQ ID NO:5)
P2:5’-TAACTGGGCCAAGGAAGGT-3’(SEQ ID NO:6)。
本发明还提出了按上述方法设计得到的引物,所述引物包括引物组,所述引物组为:
P3:5’-GACTCACGAAAGCCAGCTG-3’(SEQ ID NO:7)
P4:5’-AGTCAGCACGCCATACCA-3’(SEQ ID NO:8)
本发明还提出了将上述引物设计方法或引物分别在阪崎克罗诺杆菌和志贺氏菌相关基因(或区域)的扩增或检测中的应用。
本发明方法充分运用了基因组学数据以及相关方法、工具,能够在目标序列,(可以是特定基因或区域、也可以是全基因组)上高效率地进行引物设计,获得具有高通用性和高特异性的引物,其可靠性超过传统的仅针对特定基因序列的引物设计方法。本发明可以广泛应用于PCR、实时定量PCR、等温扩增,包括LAMP,以及其它核酸扩增反应类型中的引物设计。
附图说明
图1本发明引物设计方法的流程图。
图2实施例1的阪崎克罗诺杆菌LAMP引物的位置及正负链关系示意图。
图3利用实施例1设计的阪崎克罗诺杆菌LAMP引物进行核酸扩增反应后产物的电泳图。
图4实施例2的志贺氏菌PCR引物的位置、正负链关系示意图。
图5利用实施例2的志贺氏菌PCR引物进行核酸扩增反应后产物的电泳图。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
实施例1阪崎克罗诺杆菌(Cronobacter sakazakii)检测用LAMP引物的设计
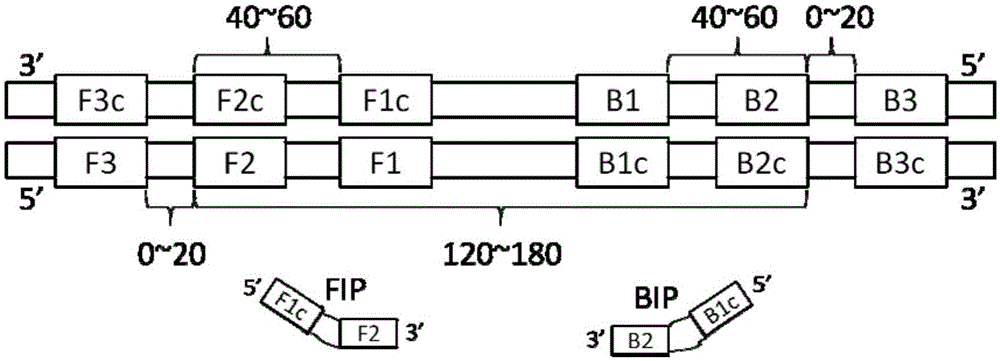
LAMP是荣研化学株式会社研发的核酸扩增方法,LAMP引物一般由来自基因组6个不同位置(分别为F3、F2、F1c、B1c、B2和B3)序列的4条单独的 引物(F3,B3,F2和F1c组成的FIP,B2和B1c组成的BIP)组成。
本发明针对阪崎克罗诺杆菌设计具有通用性和特异性的LAMP引物的方法,包括如下步骤:
(1)初始化:
使用2013年8月5号从NCBI的FTP上下载的有完整基因组序列的所有细菌和古菌的数据,共2,599个全基因组序列。设定集合G_1,其包含目标基因组序列(即,阪崎克罗诺杆菌的3个全基因组序列),其GI号分别为156932229、449306535和389839000,所设计引物应当能够检测出对应的3株阪崎克罗诺杆菌,即具有通用性;设定集合G_2,其包含非目标基因组序列(即,除阪崎克罗诺杆菌的前述3个目标基因组序列之外的2596个全基因组序列),所设计的引物应当不能够检测到除阪崎克罗诺杆菌之外的细菌和古细菌,即,具有特异性。
(2)设计单条引物:
根据LAMP引物的特性及本发明的需求,设定引物的GC含量、Tm值、3’端稳定性、5’端稳定性(见表2)及引物长度(18~25bp)等特性参数,同时设定单条引物不能产生发卡结构、自身不能发生相互作用等条件,以GI号为156932229的阪崎克罗诺杆菌ATCC BAA-894的全基因组序列作为单条引物设计的候选序列,计算得出符合上述设定条件的单条引物。并记录每条引物在156932229基因组序列上的位置、正负链信息(即来源于正链还是负链)以及引物的长度等信息。
表2 LAMP反应中单条引物的特性要求
(3)引物序列比对:
使用比对软件Bowtie,将上一步设计出的单条引物分别与集合G_1中的所 有目标基因组和集合G_2中的所有非目标基因组进行序列比对。为了确保获得的引物的通用性,当该单条引物与集合G_1中的目标序列进行比对时,使用参数设置“-a -n 0”,即要求该单条引物与所有目标序列完全匹配。为了确保获得的引物的特异性,当该单条引物与集合G_2中的非目标序列进行比对时,使用参数“-a -n 3”,即,该单条引物与所有非目标序列不超过3个错配。比对结束后,获得符合上述比对条件的单条引物,并记录每条引物在集合G_1和集合G_2中的每个基因组上的位置、正负链信息(即来源于正链还是负链)等信息。
(4)单条引物组合:
从上述步骤中产生的候选的单条引物F3、F2、F1c、B1c、B2和B3,按照图2中的条件设定这六条单条引物之间的位置以及在正负链上的关系,将单条引物组合成引物组。单条引物组合成引物组可按通用方法操作。
(5)引物组的检测:
首先对引物组进行物理化学性质检测,包括:①引物组在目标序列或者非目标序列上能否满足引物之间的位置关系、正负链关系;②引物组的GC含量、Tm值等与其扩增区域的GC含量是否满足相互关系;③每一条引物与其它单条引物之间是否存在相互作用。
其次,对引物组进行通用性检测和判定,即检测该引物组能否应用在集合G_1中的多个目标序列。该引物组能应用于集合G_1中的越多的目标序列,通用性越好。
接着,对引物组进行特异性检测和判定,即检测该引物组能否用于集合G_2中的任意一个非目标序列。若不能用于集合G_2中的任意一个非目标序列,则特异性检测结果为“通过”,若能用于集合G_2中的一个或多个非目标序列,则特异性检测结果为“未通过”。
通过上述物理化学性质、通用性及特异性三项检测的,则该引物组即为符合条件的引物组。若未通过上述物理化学性质、通用性及特异性三项检测,则放弃该引物组。继续或重新筛选,直至获得符合条件的引物组。
(6)输出符合条件的引物组:
输出前步骤中获得的能够同时通过物理化学性质检测、通用性和特异性检测的引物组中每一条引物的位置、长度、正负链信息(即来源于正链还是负链)、 以及该引物组的通用性情况(即可以用于哪些目标序列上)等信息。系统将符合条件的引物组按照通用性程度排序,通用性越强,排名越靠前,系统按此顺序输出符合条件的引物组。其中,引物组的数量可预先设定,在本实施例中引物组的数量预先设定为5。
例如,经过运行程序在AT富集区设计出LAMP反应引物组5个,随机选择一个引物组进行有效性验证。所述引物组的序列为:
F3:5’-GGATTAGGAAATATACGACGC-3’(SEQ ID NO:1)
FIP:5’-AGCAAAACCCATTGGTACCTTATACTGTCTATGGGCACAA-3’(SEQ ID NO:2)
BIP:5’-TGAATATGATCCCCCGGCTTCTGGGAGGAAATATCGCTAA-3’(SEQ ID NO:3)
B3:5’-AGCATATATGATACGGGGAA-3’(SEQ ID NO:4)
所用的LAMP反应体系如下(反应总体积为25μL):1×Thermopol反应缓冲液,6mmol/L Mg2+,1.6mmol/L dNTP,0.8μmol/L的FIP和BIP引物,0.3μmol/L的F3和B3引物,8U Bst DNA聚合酶,0.8mol/L甜菜碱,和不同浓度的DNA模板(1ng~1pg)。
当使用阪崎克罗诺杆菌(编号为CICC 21560)DNA为模板时,采用设计的引物组成功地进行了扩增,扩增产物的电泳图如图3所示,泳道M为DNA分子量标准DL2000,泳道1~4分别为DNA模板量为1ng、100pg、10pg和1pg的阪崎克罗诺杆菌的LAMP扩增产物,呈现出LAMP反应特征性的梯形条带,泳道5为以灭菌水作阴性对照模板扩增后的产物,不呈现LAMP反应特征性的梯形条带。
根据该引物组在GI号为156932229的阪崎克罗诺杆菌(Cronobacter sakazakii)ATCC BAA-894上的位置,将F3到B3之间的扩增区域序列(包含F3和B3)与数据库中阪崎克罗诺杆菌的另外两个全基因组序列(GI分别为389839000和449306535)进行Blast比对,结果显示,引物区序列均完全匹配,表明本实施例中的上述引物组可以扩增目前已有的3株阪崎克罗诺杆菌,通用性很好。
对上述引物组进行特异性检测。首先,从理论上进行分析,将该引物组与其它所有非阪崎克罗诺杆菌的基因组进行Bowtie比对(将参数设置为“-a -n 3”, 即允许不超过3个碱基不匹配),发现上述引物组的序列在非阪崎克罗诺杆菌的基因组中不存在。此外,从对28株非阪崎克罗诺杆菌和1株阪崎克罗诺杆菌菌株进行的扩增实验结果(见表3)可以看出,使用该引物组仅有阪崎克罗诺杆菌菌株扩增结果为阳性,而其它非阪崎克罗诺杆菌菌株均为阴性。因此,上述引物组具有很好的特异性。
综合上述实验及理论分析可知,根据本发明方法设计出的阪崎克罗诺杆菌的LAMP引物具有很好的通用性和特异性,满足设计的要求。
表3引物特异性检测的实验结果
注:a)CGMCC:中国普通微生物菌种保藏中心,CICC:中国工业微生物菌种保藏管理中心,CMCC:中国医学细菌菌种保藏管理中心。b)+:阳性结果,-:阴性结果。
实施例2志贺氏菌检验用PCR引物的设计
本发明针对志贺氏菌(Shigella spp.)设计具有通用性和特异性的PCR引物,步骤如下:
(1)初始化:
使用2013年8月5号从NCBI的FTP上下载的有完整基因组序列的所有细菌和古菌的数据,共2599个全基因组序列。集合G_1中包含目标基因组序列(即,志贺氏菌的9个全基因组序列),集合G_2中包含非目标基因组序列(即,除前述集合G_1中的9个全基因组序列之外的2590个全基因组序列)。
(2)设计单条引物:
PCR引物来源于基因组中的2个位置(引物P1和引物P2)。根据PCR引物的特性以及本实施例的需求,设定引物的GC含量、Tm值、3’端稳定性、5’端稳定性(见表4)及引物长度(18~25bp)等特性参数,且规定单条引物不能产生发卡结构、自身不能发生相互作用等条件,以GI号为110804074的福氏志贺氏菌(Shigella flexneri 5str.8401)的全基因组序列作为单条引物设计的候选序列,计算得出符合条件的单条引物。并记录每一个单条引物在110804074基因组序列上的位置、正负链信息(即来源于正链还是负链)以及引物的长度等信息。
表4 PCR反应中单条引物的特性要求
(3)引物序列比对:
使用比对软件Bowtie,将上一步设计出的单条引物分别与集合G_1中的目标基因组序列和集合G_2中的非目标基因组进行序列比对。为了保证引物的通用性,当该单条引物与集合G_1中的目标序列进行比对时,使用参数设置“-a -n 0”,即要求该单条引物与目标序列完全匹配;为了保证引物的特异性,当该单条引物与集合G_2中的非目标序列进行比对时,使用参数设置“-a -n 3”,即允许该单条引物与非目标序列有不超过3个错配。比对结束后,记录每一个单条引物在集合G_1和集合G_2中的每个基因组上的位置、正负链信息(即来源于正链还是负链)。
(4)单条引物组合:
从上述步骤中产生的候选的单条引物中挑选引物P1和引物P2,并按照图4中的条件设定2条单条引物之间的位置以及在正负链上的关系,将单条引物组合成引物组。
(5)引物的检测:
首先对引物组进行物理化学性质检测,包括:①引物组在目标序列或者非目标序列上能否满足引物之间的位置关系、正负链关系;②引物组的GC含量、Tm值等与其扩增区域的GC含量是否满足相互关系;③每一条引物与其它单条引物之间是否存在相互作用。
其次,对引物组进行通用性检测和判定,即检测该引物组能否应用在集合G_1中的9个目标序列。该引物组能应用于集合G_1中的越多的目标序列,通用性越好。
接着,对引物组进行特异性检测和判定,即检测该引物组能否用于集合G_2中的任意一个非目标序列,若不能用于集合G_2中的任意一个非目标序列,则特异性检测结果为“通过”,若能用于集合G_2中的一个或多个非目标序列,则特异性检测结果为“未通过”。
通过上述物理化学性质、通用性及特异性三项检测的,则该引物组即为符合条件的引物组。若未通过上述物理化学性质、通用性及特异性三种检测,则放弃该引物组。继续或重新筛选,直至获得符合条件的引物组。
(6)输出符合条件的引物组:
输出前步骤中获得的能够同时通过物理化学性质检测、通用性和特异性检测的引物组中每一条引物的位置、长度、正负链信息(即来源于正链还是负链)、以及该引物组的通用性情况(即可以用于哪些目标序列上)等信息。系统将符合条件的引物组按照通用性程度排序,通用性越强,排名越靠前,系统按此顺序输 出符合条件的引物组。其中,引物组的数量可预先设定,在本实施例中引物组的数量预先设定为5。
例如,经过运行程序在AT富集区设计出PCR反应所需要的引物组5个,随机选择一个引物组进行有效性验证。所述引物组的序列为:
P1:5’-GGAGCCTGCTCAGTCTGTT-3’(SEQ ID NO:5)
P2:5’-TAACTGGGCCAAGGAAGGT-3’(SEQ ID NO:6)
所用的PCR反应体系(反应总体积为25μL)为:1×PCR反应缓冲液(含Mg2+),0.1mmol/L dNTP,0.8μmol/L的P1和P2引物和0.1U/μL Taq DNA聚合酶和50ng/μL DNA模板。所用的反应温度条件为:95℃预变性3min;95℃变性30s,不同温度(51℃、53℃、55℃、57℃和59℃)退火30s,72℃延伸30s,循环30次;最后72℃延伸7min;16℃保存。
当使用志贺氏菌(编号为CGMCC1.1868,为福氏志贺氏菌)DNA为模板时,采用设计的引物组成功进行了扩增,扩增产物的电泳图如图5所示,泳道M为100bp梯度DNA分子量标准DNAMarker I(100bp~600bp),泳道1~4分别为退火温度为51℃、53℃、55℃、57℃和59℃的志贺氏菌的PCR扩增产物,其片段大小约为240bp,与预期片段大小(242bp)一致,泳道5为以灭菌水作阴性对照模板扩增后的产物,没有条带。
通过将该引物组的P1和P2序列与所有细菌基因组的数据库进行Bowtie比对(允许不超过3个错配),在理论上对该引物组的特异性进行分析,结果发现两条引物仅在志贺氏菌的9个基因组里同时出现,而在所有其它细菌基因组里均不能同时出现,表明采用本发明方法设计的该引物组具有很好的特异性。
综合上述实验及理论分析可知,根据本发明方法设计出的志贺氏菌的PCR引物具有很好的通用性和特异性,满足设计的要求。
实施例3阪崎克罗诺杆菌(Cronobacter sakazakii)OmpA基因扩增用PCR引物的设计
利用本发明设计用以扩增阪崎克罗诺杆菌OmpA基因的PCR引物,步骤如下:
(1)初始化:
使用2013年8月5号从NCBI的FTP上下载的三株阪崎克罗诺杆菌的完整 基因组序列(GI号分别为156932229、449306535和389839000)。设定集合G_1,其包含三株阪崎克罗诺杆菌的OmpA基因序列;设定集合G_2,其包含三株阪崎克罗诺杆菌基因组中除OmpA基因之外的其它序列。
(2)设计单条引物:
根据PCR引物的特性以及本实施例的需求,设定引物(P3和P4)的GC含量、Tm值、3’端稳定性、5’端稳定性(见表4)及引物长度(18~25bp)等特性参数,且规定单条引物不能产生发卡结构、自身不能发生相互作用等条件,以GI号为156932229的阪崎克罗诺杆菌的OmpA基因序列作为单条引物设计的候选序列,计算得出符合条件的单条引物。并记录每一个单条引物在156932229基因组序列上的位置、正负链信息(即来源于正链还是负链)以及引物的长度等信息。
(3)引物序列比对:
使用比对软件Bowtie,将上一步设计出的单条引物分别与集合G_1中的目标基因组序列和集合G_2中的非目标基因组进行序列比对。为了保证引物的通用性,当该单条引物与集合G_1中的目标序列进行比对时,使用参数设置“-a -n 0”,即要求该单条引物与目标序列完全匹配;为了保证引物的特异性,当该单条引物与集合G_2中的非目标序列进行比对时,使用参数设置“-a -n 3”,即允许该单条引物与非目标序列有不超过3个错配。比对结束后,记录每一个单条引物在集合G_1和集合G_2中的每个基因组上的位置、正负链信息(即来源于正链还是负链)。
(4)单条引物组合:
从上述步骤中产生的候选的单条引物中挑选引物P3和引物P4,并按照图4中的条件设定2条单条引物之间的位置以及在正负链上的关系,将单条引物组合成引物组。
(5)引物的检测:
首先对引物组进行物理化学性质检测,包括:①引物组在目标序列或者非目标序列上能否满足引物之间的位置关系、正负链关系;②引物组的GC含量、Tm值等与其扩增区域的GC含量是否满足相互关系;③每一条引物与其它单条引物之间是否存在相互作用。
其次,对引物组进行通用性检测和判定,即检测该引物组能否应用在集合G_1中的9个目标序列。该引物组能应用于集合G_1中的越多的目标序列,通用性越好。
接着,对引物组进行特异性检测和判定,即检测该引物组能否用于集合G_2中的任意一个非目标序列,若不能用于集合G_2中的任意一个非目标序列,则特异性检测结果为“通过”,若能用于集合G_2中的一个或多个非目标序列,则特异性检测结果为“未通过”。
通过上述物理化学性质、通用性及特异性三项检测的,则该引物组即为符合条件的引物组。若未通过上述物理化学性质、通用性及特异性三种检测,则放弃该引物组。继续或重新筛选,直至获得符合条件的引物组
(6)输出符合条件的引物组:
输出前步骤中获得的能够同时通过物理化学性质检测、通用性和特异性检测的引物组中每一条引物的位置、长度、正负链信息(即来源于正链还是负链)、以及该引物组的通用性情况(即可以用于哪些目标序列上)等信息。系统将符合条件的引物组按照通用性程度排序,通用性越强,排名越靠前,系统按此顺序输出符合条件的引物组。其中,引物组的数量可预先设定(在本实施例中引物组的数量预先设定为5)。
例如,经过运行程序在GC含量正常区设计出PCR反应所需要的引物组5个,随机选择一个引物组进行有效性验证。所述引物组的序列为:
P3:5’-GACTCACGAAAGCCAGCTG-3’(SEQ ID NO:7)
P4:5’-AGTCAGCACGCCATACCA-3’(SEQ ID NO:8)
所用的PCR反应体系(反应总体积为25μL)为:1×PCR反应缓冲液(含Mg2+),0.1mmol/L dNTP,0.8μmol/L的P3和P4引物,0.1U/μL Taq DNA聚合酶和50ng/μL DNA模板。所用的反应温度条件为:95℃预变性3min;95℃变性30s,55℃退火30s,72℃延伸30s,循环30次;最后72℃延伸7min;16℃保存。
当使用阪崎克罗诺杆菌(编号为CICC 21560)DNA为模板时,采用设计的引物组进行了扩增,电泳分析结果表明,扩增产物与预期片段大小(230bp)一致。
因此,根据本发明方法设计出的阪崎克罗诺杆菌OmpA基因的PCR引物能够满足设计的要求。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种引物组及其在扩增siv/shiv基因组中的应用与试剂盒的制作方法
- 检测f8基因突变的扩增引物、试剂盒及方法
- 拟茎点霉rpb2基因扩增引物及其设计方法和应用
- 一种马来穿山甲线粒体Cox3基因序列的扩增引物及鉴定方法
- 拟茎点霉gpdh基因扩增引物及其设计方法和应用
- 甲型流感病毒全基因组扩增方法及使用的复合引物和试剂盒的制作方法
- 用于检测2型糖尿病视网膜病变的miR-2116-5p分子标记物及其扩增引物和应用
- 用于检测2型糖尿病视网膜病变的miR-3197分子标记物及其扩增引物和应用
- 一种用于鱼类线粒体全基因组扩增的引物、试剂盒及扩增方法
- 用Alu重复序列基因扩增引物且无需DNA抽提的检测血浆游离DNA的定量PCR方法