CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与流程
本发明涉及基因工程技术领域,尤其涉及基因敲除技术领域,具体涉及CRISPR-Cas9特异性敲除猪SLA-3基因的方法及用于特异性靶向SLA-3基因的sgRNA。
背景技术:
器官移植是治疗器官衰竭疾病最有效的治疗手段。迄今为止,全球已有近百万的患者通过器官移植而延续生命。随着人口老龄化及医疗技术的进步,需要进行器官移植手术的病人越来越多,但供体器官的短缺严重制约了器官移植手术的开展。以肾脏移植为例,我国每年需要进行肾移植的患者多达30万,而可用于移植的捐献肾脏不超过1万例,大部分患者死于肾衰竭。依靠死后器官捐献已不能满足器官移植的需要。通过基因工程改造其他物种,以提供合适于人体移植的器官,成为解决人类供体器官短缺问题的主要途径。
目前,根据生物安全性、生理功能指标、经济性及稀有物种保护等多方面评价,猪成为了最为理想的异种器官来源。但猪和人之间存在巨大的差异,直接将猪的器官移植到人会产生强烈的免疫排斥反应。因此,通过基因工程对猪进行改造,以产生适合于人体移植的器官,成为异种移植的终极目标。
猪白细胞抗原(swine leukocyte antigen,SLA)I类分子是代表基因分子遗传特征的重要功能基因。分别称为SLA-1(PD1)、SLA-2(PD14)和SLA-3(PD7),也分别称为SLA-C、SLA-B和SLA-A。经典的SIJA基因主要高表达于免疫系统和消化系统,尤其在免疫系统中,脾、胸腺、支气管淋巴结等组织富含免疫细胞。人CD4T细胞通过间接的抗原递呈途径识别猪的异种抗原,且主要为SLA I类分子所识别。因此,需要消除猪细胞表面的SLA I类分子以减少异种供体器官的免疫原性。而准确高效的敲除猪SLA-3基因,是消除SLA I类分子引起的免疫排斥的关键步骤。
目前,常见的基因敲除技术包括同源重组(Homologus Recombination,HR)技术、类转录激活效应子核酸酶(Transcription Activator-Like Effector Nuclease,TALEN)技术、锌指核酸酶(Zinc-Finger Nuclease,ZFN)技术以及最近发展的规律成簇间隔短回文重复(Clustered Regularly Interspaced Short Palindromic Repeat,CRISPR)技术。HR技术由于重组效率低下(效率大约只有10-6),对突变体的筛选工作非常耗时和低效,已逐渐被取代。TALEN技术和ZFN技术的切割效率一般能达到20%,但都需要构建可以识别特定序列的蛋白质模块,前期工作繁琐费时。ZFN技术的模块设计较为复杂且有较高的脱靶率,其应用有限。
CRISPR是一种源于原核生物的后天免疫系统,该系统执行干扰功能的复合物由蛋白质Cas和CRISPR-RNA(crRNA)组成。目前该系统已发现有三种类型,其中第二类Cas9系统组成简单,已被积极应用于基因工程领域。Cas9靶向切割DNA是通过两种小RNA——crRNA(CRISPR RNA)和tracrRNA(trans-activating crRNA)与靶序列互补识别的原理实现的。现在已经将两种小RNA融合成一条RNA链,简称sgRNA(single guide RNA),能够识别特定的基因序列,引导Cas9蛋白进行切割。在真核生物中,DNA被切断后发生非同源重组末端连接,造成移码突变,最终导致基因功能性敲除。
相比于上述3种技术,CRISPR技术操作简单、筛选效率高,能够实现精确的靶向切割。因此,通过CRISPR技术敲除SLA-3基因能够极大地提高Neu5Gc缺失细胞及基因工程猪的筛选效率。但是该路径的关键技术难题是设计并制备精确靶向的sgRNA,因为基因的靶向精确度高度依赖于sgRNA靶序列,能否成功设计出精确靶向的sgRNA成为敲除目的基因的关键技术问题,本发明意在解决该技术问题从而为敲除SLA-3基因提供坚实的基础。
技术实现要素:
本发明的目的在于提供CRISPR-Cas9特异性敲除猪SLA-3基因的方法及用于特异性靶向SLA-3基因的sgRNA。
根据本发明的第一方面,本发明提供在CRISPR-Cas9特异性敲除猪SLA-3基因中用于特异性靶向SLA-3基因的sgRNA,该sgRNA具有以下特点:
(1)该sgRNA在SLA-3基因上的靶序列符合5’-N(20)NGG-3’的序列排列规则,其中N(20)表示20个连续的碱基,其中每个N表示A或T或C或G,符合规则的靶序列可以位于正义链或反义链;
(2)该sgRNA在SLA-3基因上的靶序列位于SLA-3基因的N端的5个外显子编码区,或序列的主要部分位于SLA-3基因的N端的5个外显子,其余部分跨越与相邻内含子的交界,位于相邻内含子;
(3)该sgRNA在SLA-3基因上的靶序列是唯一的。
作为本发明的优选方案,上述靶序列为序列表中SEQ ID NO:1~115中任一条序列所示的序列。
作为本发明的优选方案,上述靶序列为序列表中SEQ ID NO:4、5或12所示的序列。
根据本发明的第二方面,本发明提供CRISPR-Cas9特异性敲除猪SLA-3基因的方法,该方法包括如下步骤:
(1)在第一方面所述的sgRNA的靶序列的5’-端加上用于形成粘性末端的序列,合成得到正向寡核苷酸序列;在第一方面所述的sgRNA的靶序列对应的互补序列的两端加上合适的用于形成粘性末端的序列,合成得到反向寡核苷酸序列;将合成的正向寡核苷酸序列与反向寡核苷酸序列退火、复性,形成具有粘性末端的双链寡聚核苷酸;
(2)将上述双链寡聚核苷酸连入线性化的携带Cas9基因的表达载体,得到携带含相应靶序列的sgRNA寡聚核苷酸和Cas9基因的表达载体,转化感受态细菌,筛选鉴定出正确的阳性克隆,并对阳性克隆摇菌、提取质粒;
(3)用上述携带有sgRNA寡聚核苷酸和Cas9基因的表达载体、包装质粒和包装细胞系包装出同时携带靶向SLA-3基因的sgRNA和Cas9的假型慢病毒;
(4)使用上述假型慢病毒感染目的细胞,并进一步培养;然后收集被感染的目的细胞,以其基因组DNA为模板扩增包含上述靶序列的基因片段,经过变性、复性及酶切,确定SLA-3基因的敲除情况。
作为本发明的优选方案,上述表达载体为序列表中SEQ ID NO:116所示序列的载体。
作为本发明的优选方案,上述方法包括如下步骤:
(1)在第一方面所述的sgRNA的靶序列的5’-端加上CACCG序列,合成得到正向寡核苷酸序列;在第一方面所述的sgRNA的靶序列对应的互补序列的5’-端加上AAAC序列、3’-端加上C,合成得到反向寡核苷酸序列;将合成的正向寡核苷酸序列与反向寡核苷酸序列退火、复性,形成具有粘性末端的双链寡聚核苷酸;
(2)将上述双链寡聚核苷酸连入如序列表中SEQ ID NO:116所示序列的表达载体lentiCRISPR v2经BsmB I限制性内切酶酶切得到的线性化载体,得到携带sgRNA寡聚核苷酸的重组表达载体lentiCRISPR v2-SLA-3,转化感受态细菌,筛选鉴定出正确的阳性克隆,并对阳性克隆摇菌、提取质粒;
(3)用上述表达载体lentiCRISPR v2-SLA-3、包装质粒和包装细胞系包装出同时携带靶向SLA-3基因的sgRNA和Cas9的假型慢病毒;
(4)使用上述CRISPR假型慢病毒感染目的细胞,并进一步培养;然后收集被感染的目的细胞,以其基因组DNA为模板扩增包含上述靶序列的基因片段,经过变性、复性及酶切,确定SLA-3基因的敲除情况。
作为本发明的优选方案,上述包装质粒为质粒pLP1、质粒pLP2和质粒pLP/VSVG;上述包装细胞系为HEK293T细胞。
作为本发明的优选方案,上述目的细胞为猪PIEC细胞。
作为本发明的优选方案,上述以其基因组DNA为模板扩增包含上述靶序列的基因片段,经过变性、复性及酶切,确定SLA-3基因的敲除情况,具体为:
(a)以感染病毒的目的细胞的基因组DNA为模板,用SLA-3基因的上下游引物扩增包含上述sgRNA的靶序列的SLA-3基因片段,同时用相同引物扩增未感染病毒的野生型细胞的基因组DNA;
(b)纯化上述扩增到的SLA-3基因片段,然后将来自感染病毒的目的细胞的SLA-3基因片段与来自野生型细胞的SLA-3基因片段等量混合,加热变性、复性,形成杂交DNA分子;
(c)用Cruiser酶切割复性后的杂交DNA分子;
(d)电泳检测酶切产物,检测靶序列介导的SLA-3基因敲除效果。
根据本发明的第三方面,本发明提供在CRISPR-Cas9特异性敲除猪SLA-3基因的方法中用到的重组表达载体lentiCRISPR v2-SLA-3,该重组表达载体的骨架载体的序列如序列表中SEQ ID NO:116所示;所携带的靶序列如第一方面的sgRNA的靶序列,优选序列表中SEQ ID NO:4、5或12所示的靶序列。
根据本发明的第四方面,本发明提供如第一方面所述的sgRNA或第三方面所述的重组表达载体lentiCRISPR v2-SLA-3在CRISPR-Cas9特异性敲除猪SLA-3基因的方法中的用途。
本发明的针对CRISPR-Cas9特异性敲除猪SLA-3基因,成功地找到特异性靶向SLA-3基因的sgRNA,将本发明的sgRNA用于CRISPR-Cas9特异性敲除猪SLA-3基因的方法中,能够快速、精确、高效、特异性地敲除猪SLA-3基因,有效地解决构建SLA-3基因敲除猪周期长和成本高的技术问题。
附图说明
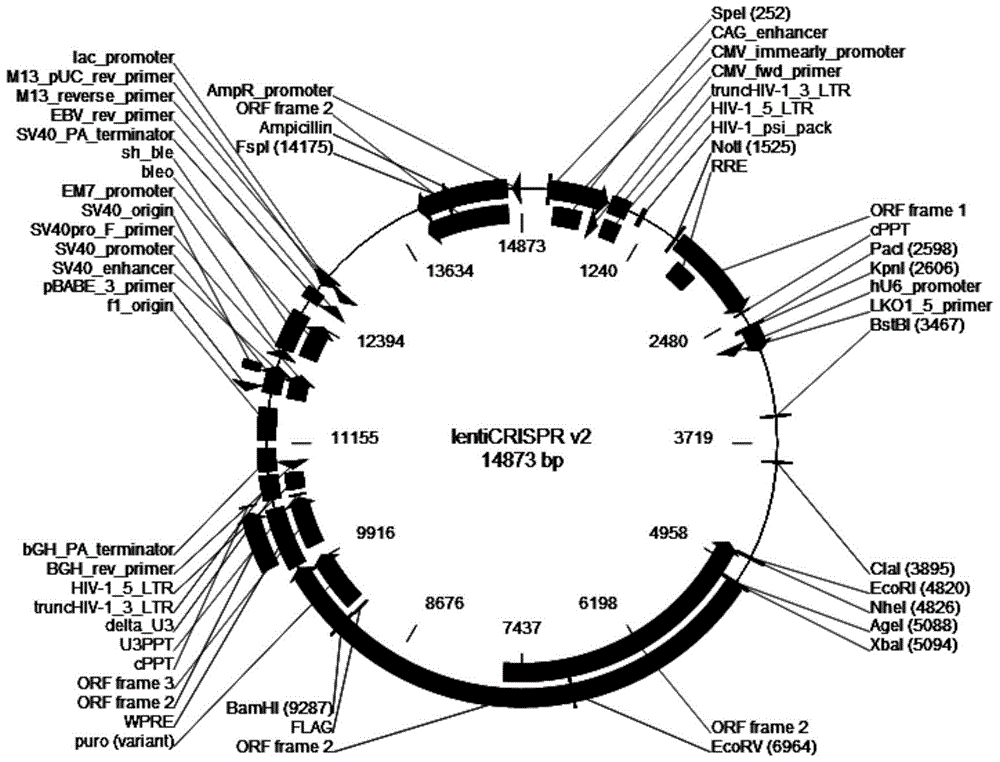
图1为本发明实施例中使用的载体质粒lentiCRISPR v2的质粒图谱;
图2为本发明实施例中使用的包装质粒pLP1的质粒图谱;
图3为本发明实施例中使用的包装质粒pLP2的质粒图谱;
图4为本发明实施例中使用的包装质粒pLP/VSVG的质粒图谱;
图5为本发明实施例中酶切验证靶序列的基因敲除效果的电泳检测结果图,其中M表示DNA Marker,4、5和12分别表示表1中第4号、第5号和第12号靶序列对SLA-3基因的靶向切割效果,WT表示未经过病毒感染和Cas9切割的野生型细胞的PCR产物Cruiser酶切检测结果,箭头处表示经Cruiser酶切割得到的小片段。
具体实施方式
下面结合附图和具体实施例对本发明的技术方案做进一步说明。这些附图和具体实施例不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段,所用原料均为市售商品。
以下实施例中涉及的试验材料和试剂:lentiCRISPR v2质粒购自Addgene公司,包装质粒pLP1、pLP2和pLP/VSVG购自Invitrogen公司,包装细胞系HEK293T细胞购自美国模式培养物集存库(ATCC),PIEC细胞购自中国科学院细胞库,DMEM培养基、Opti-MEM培养基和胎牛血清FBS购自Gibco公司,Lipofectamine 2000购自Invitrogen公司。
以下实施例中未作具体说明的分子生物学实验方法,均参照《分子克隆实验指南》(第三版)J.萨姆布鲁克一书中描述的具体方法进行,或者按照试剂盒和产品说明书进行。
本发明的概括性的技术方案包括以下五个部分:
一、Sus scrofa(猪)SLA-3基因sgRNA靶序列的选择和设计
1.SLA-3基因的sgRNA靶序列选择:
在SLA-3基因外显子区寻找合适的20bp寡核苷酸序列作为靶序列。
2.SLA-3基因的sgRNA靶序列设计:
将上述靶序列及互补序列分别添加接头,形成正向寡核苷酸序列和反向寡核苷酸序列。
二、构建SLA-3基因的CRISPR载体
1.合成上述正向寡核苷酸序列和反向寡核苷酸序列,复性形成具有粘性末端的双链DNA片段(即双链靶序列寡聚核苷酸)。
2.构建CRISPR-sgRNA表达载体:
将上述双链DNA片段构建至目标载体(如lentiCRISPR v2,其质粒图谱如图1所示),形成如lentiCRISPR v2-SLA-3的慢病毒CRISPR载体。
三、获得表达SLA-3sgRNA的假型慢病毒
利用包装质粒、包装细胞系与慢病毒CRISPR载体生产表达SLA-3sgRNA的CRISPR假型慢病毒。
四、感染目的细胞并检测SLA-3基因敲除效果
1.慢病毒感染目的细胞:
将如lentiCRISPR v2-SLA-3的假型慢病毒加入目的细胞培养基进行感染并进一步培养。
2.检测SLA-3基因敲除效果:
收集目的细胞,以基因组DNA为模板扩增包含靶序列的基因片段,经过变性、复性及酶切,确定SLA-3基因的敲除情况。
五、SLA-3基因敲除单克隆的挑选和鉴定
1.对于有确定敲除效果的目的细胞群,通过稀释和单克隆培养,分离出若干单细胞来源的细胞株。
2.鉴定单克隆的SLA-3敲除情况。
以下通过实施例详细说明本发明的技术方案及其有益效果。
实施例一、Sus scrofa(猪)SLA-3基因sgRNA靶序列的选择和设计
靶序列决定了sgRNA的靶向特异性和诱导Cas9切割目的基因的效率。因此,高效特异的靶序列选择和设计是构建sgRNA表达载体的前提。
1.SLA-3基因的sgRNA靶序列选择
针对SLA-3基因,在靶序列选择上应该遵循下列原则:
(1)在SLA-3基因外显子编码区寻找符合5’-N(20)NGG-3’规则的靶序列,其中N(20)表示20个连续的碱基,其中每个N表示A或T或C或G,符合规则的靶序列可以位于正义链或反义链;
(2)选择靠近N端的5个外显子编码区序列,这样的编码区序列的切割会造成SLA-3基因的功能敲除,残留截短的序列不会形成有功能的蛋白;
(3)如果存在多种剪切体,则在共有外显子编码区进行选择,针对SLA-3基因选择靠近N端的5个外显子编码区序列即可满足该条件;
(4)利用在线序列分析工具(http://crispr.mit.edu/)分析以上靶序列在猪基因组中的同源情况,舍弃存在显著同源序列的靶序列,根据评分进一步挑选,所挑选的靶序列在SLA-3基因上是唯一的。
基于以上原则,选择出表1所示的靶序列集合。
表1 靶序列集合
2.SLA-3基因的sgRNA靶序列设计:
(1)以lentiCRISPR v2质粒作为表达载体,根据lentiCRISPR v2质粒的特点,在上述N(20)靶序列的5’-端添加CACCG序列,形成正向寡核苷酸序列:
5’-CACCGNNNNNNNNNNNNNNNNNNNN-3’;
(2)在上述N(20)靶序列的反向互补序列的两端添加序列,形成反向寡核苷酸序列:
5’-AAACNNNNNNNNNNNNNNNNNNNNC-3’;
正向寡核苷酸序列和反向寡核苷酸序列可以互补形成具有粘性末端的双链DNA片段:
5’-CACCGNNNNNNNNNNNNNNNNNNNN-3’
3’-CNNNNNNNNNNNNNNNNNNNNCAAA-5’。
实施例二、构建SLA-3基因的sgRNA表达载体
1.合成DNA插入片段
(1)合成上述设计的正向和反向寡核苷酸序列
寡核苷酸序列可以由商业化的公司(如Invitrogen公司)根据提供的序列具体合成。本实施例及以下实施例研究了表1中所列的第4号、第5号和第12号序列所示的靶序列对SLA-3基因的敲除效果。
第4号靶序列对应的正向寡核苷酸序列和反向寡核苷酸序列如下:
5’-CACCGGGCCCTGACTGGTACCCGGG-3’(SEQ ID NO:117);
5’-AAACCCCGGGTACCAGTCAGGGCCC-3’(SEQ ID NO:118)。
第5号靶序列对应的正向寡核苷酸序列和反向寡核苷酸序列如下:
5’-CACCGGCCCTGACTGGTACCCGGGA-3’(SEQ ID NO:119);
5’-AAACTCCCGGGTACCAGTCAGGGCC-3’(SEQ ID NO:120)。
第12号靶序列对应的正向寡核苷酸序列和反向寡核苷酸序列如下:
5’-CACCG CCTGGCCCTGACTGGTACCC-3’(SEQ ID NO:121);
5’-AAACGGGTACCAGTCAGGGCCAGGC-3’(SEQ ID NO:122)。
将对应的正向和反向寡核苷酸序列退火、复性,形成具有粘性末端的双链DNA片段。
反应体系(20μL)如下所示:
正向寡核苷酸(10μM):1μL
反向寡核苷酸(10μM):1μL
10×PCR buffer:2μL
ddH2O:16μL
将上述反应体系放入PCR仪,并按以下程序进行反应。
反应程序:
95℃,5min;
80℃,5min;
70℃,5min;
60℃,5min;
50℃,5min;
自然降至室温。
2.构建sgRNA表达载体
(1)利用BsmB I限制性内切酶酶切目标载体lentiCRISPR v2质粒(其序列如序列表中SEQ ID NO:116所示)。
按照以下反应体系进行配制:
LentiCRISPR v2质粒:1μg
10×酶切buffer:2μL
BsmB I限制性内切酶:2μL
补充ddH2O至总体积20μL
将酶切反应体系置于37℃反应4h。
(2)电泳分离并纯化载体片段
酶切结束后,将酶切混合物通过琼脂糖凝胶电泳进行分离,选择载体片段(约12kb)进行切割,并通过DNA凝胶回收柱进行回收。
(3)将合成的双链DNA片段与载体主片段进行连接并转化大肠杆菌
将复性得到的双链DNA片段与回收得到的载体片段进行连接反应,按照以下反应体系进行配制:
LentiCRISPR v2载体片段:100ng
双链DNA片段:200ng
T4连接酶:1μL
T4连接反应buffer:1μL
补充ddH2O至总体积10μL
将连接混合物置于25℃反应2h。
反应结束后将连接混合物转化大肠杆菌DH5α菌株:向连接混合物中加入100μL大肠杆菌DH5α感受态细胞,冰上孵育30min;将混合物放入42℃水浴,热激90s后放入冰上冷却;向混合物加入100μL LB培养基,37℃摇床培养20min;将混合物涂Amp LB平板,37℃培养14h。
(4)鉴定正确的转化克隆
从Amp LB平板上挑选若干菌落进行扩大培养,提取质粒进行酶切鉴定。挑选可能正确的克隆进行测序,验证插入序列是否正确。对于正确的lentiCRISPR v2-SLA-3载体克隆进行保种。
实施例三、获得表达SLA-3sgRNA的假型慢病毒
1.材料准备
扩增并抽提包装质粒pLP1、pLP2和pLP/VSVG(购自Invitrogen,其图谱分别如图2、图3和图4所示);扩增并抽提载体质粒lentiCRISPR v2-SLA-3;培养包装细胞系HEK293T细胞(购自ATCC);DMEM培养基、Opti-MEM培养基和胎牛血清FBS(购自Gibco);Lipofectamine2000(购自Invitrogen);HEK293T细胞培养于含5%CO2的37℃培养环境中,培养基为含10%FBS的DMEM培养基。
2.转染和病毒包装
第一天:将包装细胞系HEK293T传代至10cm dish,约30%融合度;
第二天:在HEK293T达到80%融合度时按照下列配方进行转染:
配制混合物1,包含:
lentiCRISPR v2-SLA-3:6μg
pLP1:6μg
pLP2:6μg
pLP/VSVG:3μg
Opti-MEM:500μL。
配制混合物2,包含:
Lipofectamine 2000:30μL
Opti-MEM:500μL。
静置5min后,将混合物1和混合物2混匀成转染混合物,静置20min。
将HEK293T培养基换为无血清DMEM培养基,加入转染混合物,37℃培养8h后换为20%FBS的DMEM培养基,继续培养。
3.病毒收集与保存
第三天:转染48h后收集含病毒的HEK293T培养基上清,用0.45μm滤头过滤后,分装,放置-80℃保存。
实施例四、感染目的细胞并检测靶序列的敲除效果
1.材料准备
培养目的细胞系猪髋动脉血管内皮细胞PIEC(购自中国科学院细胞库);DMEM培养基和胎牛血清FBS(购自Gibco);不同靶序列(序列4、序列5和序列12)的lentiCRISPR v2-SLA-3假型慢病毒;PIEC细胞培养于含5%CO2的37℃培养环境中,培养基为含10%FBS的DMEM培养基。
2.慢病毒感染目的细胞
第一天:将目的细胞传代至6孔板,约20%融合密度。每一种病毒需要一个6孔,同时需要效率对照一个6孔。
第二天:待目的细胞约40%融合密度时加入1mL lentiCRISPR v2-SLA-3假型慢病毒上清及1mL DMEM培养基。效率对照不需要添加慢病毒。
第三天:感染24h后去除含病毒培养基,换成正常培养基,加入嘌呤霉素至终浓度2μg/mL,没有感染病毒的效率对照样品也同时加入嘌呤霉素作为对照,培养48h。
3.细胞感染效率检测和培养
第五天:未感染的效率对照细胞在嘌呤霉素的作用下应该全部凋亡(>95%)。根据感染慢病毒细胞的凋亡情况判断细胞的感染效率,通常可以达到90%以上的感染效率(凋亡率<10%)。必要时可以将病毒上清进行浓缩或梯度稀释后进行感染以达到合适的感染效率。
经过嘌呤霉素筛选后,未感染的细胞发生凋亡。将目的细胞重新传代并换为普通培养基培养48h。
4.检测SLA-3基因敲除效果
(1)设计上下游引物以扩增SLA-3基因片段,其中上下游引物序列如下所示:
CTCGGGGTCTCAGGCTCCAGGGCGG(SEQ ID NO:123)
GGGCAGGAAACAAGGGAGGG(SEQ ID NO:124)。
目的扩增片段包含sgRNA靶序列,大小为487bp。靶序列至片段两端的位置不少于100bp。
(2)收集部分目的细胞,使用promega基因组DNA试剂盒抽提基因组DNA。同时抽提野生型目的细胞的基因组DNA。
(3)以基因组DNA为模板扩增包含靶序列的SLA-3基因片段(包括感染的突变样品和野生型样品)。
扩增反应体系(20μL)如下:
上游引物(10μM):1μL
下游引物(10μM):1μL
2×PCR Mix:10μL
基因组DNA:100ng
以上述反应体系进行配制,放入PCR仪,并按下列程序进行反应。
反应程序:
95℃,3min
95℃,30s
58℃,20s
72℃,20s
72℃,3min;
其中第二步至第四步重复35个循环。
(4)电泳检测PCR产物并回收纯化
(5)将纯化后的DNA片段分别加热变性、复性,形成杂交DNA分子(包括突变样品和野生型样品)。
反应体系如下所示:
基因组PCR片段:200ng
5×反应buffer:2μL
反应体系共9μL
以上述反应体系进行配制,放入PCR仪,并按下列程序进行反应。
反应程序:
95℃,5min;
80℃,5min;
70℃,5min;
60℃,5min;
50℃,5min;
自然降至室温。
(6)用Cruiser酶切割复性后的杂交DNA(包括突变样品和野生型样品)
向经过变性、复性的反应混合物加入1μL Cruiser酶,45℃孵育20min。
(7)电泳检测酶切产物,检测靶序列介导的SLA-3基因敲除效果。
将经过酶切的DNA片段用2%的琼脂糖凝胶进行电泳分析,100V,25min。确定目的片段的切割情况,判断靶序列的基因敲除效果。
对突变DNA的切割识别基于以下原理:经过感染的细胞会表达sgRNA和Cas9。基因组DNA如果被sgRNA介导的Cas9蛋白靶向切割,经过修复后会在切割位点附近引入突变(野生型变为突变型)。由于野生型和突变型序列在该位置不匹配,以此为模板扩增出的野生型DNA与突变型DNA经过变复性形成的杂交分子会就产生局部的环形(loop)结构。而后者可以被Cruiser酶识别并切断,导致杂交DNA分子被切割成小片段。
结果如图5所示,未经过病毒感染的野生型细胞的PCR产物未检测到小片段;而序列4、序列5和序列12能够有效靶向SLA-3基因产生切割,因此检测到小片段的存在,表明序列4、序列5和序列12能够作为CRISPR-Cas9特异性敲除猪SLA-3基因的靶序列。
实施例五、SLA-3基因敲除单克隆的挑选和鉴定
1.单克隆的挑选(基于序列4、序列5和序列12的靶序列)
(1)将部分感染的目的细胞群进行传代,取100个单细胞转移至10cm dish培养。
(2)培养约10天后,有相当数量的单克隆生长到肉眼可见的水平。
(3)用移液器头刮取独立的克隆,将细胞转移至24孔板中培养,每个孔对应一个克隆。
(4)再经过约一周的培养后,有部分克隆长至足够的数量,准备做进一步的鉴定。
2.鉴定单克隆的SLA-3敲除情况
(1)收集待检的单克隆及野生型细胞,分别抽提基因组DNA。
(2)按照前述方法,分别扩增单克隆及野生型细胞的SLA-3基因片段,所扩增的基因片段包含sgRNA靶序列。
(3)将等量的单克隆PCR片段与野生型PCR片段混合,加热变性、复性,形成杂交DNA分子。
(4)用Cruiser酶切割退火后的杂交DNA,45℃孵育20min。
(5)电泳检测酶切产物,根据是否有切割片段确定单克隆是否发生有效突变。
结果显示,基于序列4所示的靶序列的lentiCRISPR v2-SLA-3假型慢病毒感染目的细胞,从100个单细胞中随机挑选的20个单克隆经Cruiser酶酶切电泳检测,其中有17个单克隆能检测到切割小片段,表明基因敲除发生,基因敲除效率能够达到85%以上,说明序列4所示的靶序列具有很高的靶向敲除SLA-3基因的作用。基于序列5所示的靶序列的lentiCRISPR v2-SLA-3假型慢病毒感染目的细胞,从100个单细胞中随机挑选的20个单克隆经Cruiser酶酶切电泳检测,其中有19个单克隆能检测到切割小片段,表明基因敲除发生,基因敲除效率能够达到95%以上,说明序列5所示的靶序列具有很高的靶向敲除SLA-3基因的作用。基于序列12所示的靶序列的lentiCRISPR v2-SLA-3假型慢病毒感染目的细胞,从100个单细胞中随机挑选的20个单克隆经Cruiser酶酶切电泳检测,其中有18个单克隆能检测到切割小片段,表明基因敲除发生,基因敲除效率能够达到90%以上,说明序列12所示的靶序列具有很高的靶向敲除SLA-3基因的作用。
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种泰国库蠓的特异基因及其分子鉴定方法
- 一种美丽库蠓特异基因及其分子鉴定方法
- 一种特异于禽类原始生殖细胞的基因转移系统及方法
- CRISPR-Cas9特异性敲除猪SALL1基因的方法及用于特异性靶向SALL1基因的sgRNA的制作方法
- CRISPR-Cas9特异性敲除猪CMAH基因的方法及用于特异性靶向CMAH基因的sgRNA的制作方法
- CRISPR-Cas9特异性敲除猪SLA-2基因的方法及用于特异性靶向SLA-2基因的sgRNA的制作方法
- 一种安宁铗蠓的特异基因及其分子鉴定方法
- CRISPR-Cas9特异性敲除猪GGTA1基因的方法及用于特异性靶向GGTA1基因的sgRNA的制作方法
- 一种蓝腹库蠓的特异基因及其分子鉴定方法
- 扬子鳄抗病毒潜力检测的ⅰ类mhc基因的特异性引物及分型方法
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 毛酸浆的分子特异性标记引物及鉴别方法与制造工艺
- 一种检测CYP2C9基因多态性的ARMS引物的制造方法与工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 脯氨酸特异性蛋白酶基因及其应用
- 一种检测水稻粒宽等位基因的特异性PCR分子标记和方法与流程
- 丝状真菌中多个基因拷贝的同时位点特异性整合的制造方法与工艺
- 一种特异性敲减人BRCC3基因慢病毒的构建及在肺癌中的应用的制造方法与工艺
- 烟粉虱MED隐种高温耐受相关基因特异性探针及其应用的制造方法与工艺
- CRISPR/SaCas9特异性敲除人CXCR4基因的方法与流程
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 用于检测烟草对tmv抗性的n基因特异性引物对、检测方法及试剂盒的制作方法