抵抗乳糖杀伤的突变体微生物的制作方法
本发明涉及产生抵抗乳糖杀伤现象的突变微生物的方法以及涉及能够通过所述方法获得的微生物。这样的工程化微生物可用于生产特殊产品(specialtyproduct),例如但不限于特殊糖类、糖脂和半乳糖基化化合物。
背景技术:
乳糖杀伤(lactosekilling)是公知且深入研究的原理,其在乳糖与另一种碳源的存在下阻碍许多生物体的生长。然而,该现象背后的确切机制是未知的,尽管诱导该现象所需的特征相当清楚。当将乳糖添加到在另一种碳源(例如但不限于甘油或蔗糖)上生长的微生物培养物中时,发生乳糖杀伤。当乳糖转运基因为可诱导的或组成型表达(33,39,75)时,也发生乳糖杀伤。最初对于在恒化器条件下用iptg和乳糖调节乳糖通透酶之表达的大肠杆菌(e.coli)观察到乳糖杀伤(28)。后来对于苜蓿根瘤菌(rhizobiummeliloti)、乳酸克鲁维酵母(kluyveromyceslactis)和运动发酵单胞菌(zymomonasmobilis)也观察到乳糖杀伤(55,70,77)。这种现象的潜在原因之一归因于细胞的乳糖转运蛋白活性的所谓“成本”,这种“成本”导致生长减少或抑制,并且还与细胞外乳糖浓度有关(34),其在工业过程中大多保持高水平以获得足够高的产物滴度和收率。然而,现有技术表明,只要在这些条件下存在乳糖转运,就应该会发生乳糖杀伤。为了解决乳糖杀伤的问题,已经提出了缺失乳糖通透酶或严重损害乳糖摄取(34)。例如,lodi等(55)缺失(敲除)了乳酸克鲁维酵母中的乳糖通透酶,发现乳糖杀伤不再发生。他们的实验过程中的自发突变进一步表明所选择的乳糖杀伤阴性菌株在其乳糖摄取方面严重受损。然而,为了有效地合成特殊产品或生物产品,乳糖摄取是必需的。因此,缺失乳糖通透酶或严重损害乳糖通透酶活性明显不是解决方案,因为产生这样的生物产品需要1)有效的乳糖摄取和2)不导致“乳糖杀伤表型”的表达盒。先前已使用用于生产基于乳糖的生物产品的乳糖通透酶,但没有解决“乳糖杀伤表型”。在过去,这个问题通过严重减少乳糖摄取(因此产生很少甚至不产生基于乳糖的特殊产品)或者通过将生长阶段与生产阶段分开以优先获得足够的生物量来解决。在该生长阶段之后,在第二阶段中添加乳糖以产生特殊产品。在该第二阶段中,不发生或强烈减少生长(59)。
因此,目前的共识是,为了避免乳糖杀伤,必须消除或严重损害乳糖摄取,因为正常的乳糖摄取将总是导致乳糖杀伤。相比之下,本发明公开了一种筛选方法以找到允许有效摄取乳糖而不经历乳糖杀伤的乳糖通透酶表达盒!
乳糖是许多生物产品,更特别是特殊糖类(14)、糖脂和半乳糖基化化合物(例如半乳糖基脂、半乳糖神经酰胺和半乳糖基化糖苷配基)的结构单元。在许多情况下,半乳糖部分用于药物的器官靶向(28)。然而,由于上述“乳糖杀伤”现象,将乳糖作为基质与其他基质组合使用不像所认为的那样明显。通常,需要多阶段生产系统、非生长耦合细胞系统(non-growingcoupledcellsystem)以避免乳糖杀伤(32,48)。
许多特殊糖类的基本结构骨架由乳糖或半乳糖单元组成。更具体地,人乳寡糖和引申的母乳寡糖(一大类糖类和寡糖)由半乳糖和乳糖单元构成(15)。这些糖类用糖部分进一步修饰,例如n-乙酰葡糖胺、n-乙酰半乳糖胺、唾液酸(例如n-乙酰神经氨酸、n-羟乙酰神经氨酸、2-酮-3-脱氧-d-甘油半乳糖壬酮糖酸……)(21),l-岩藻糖等。然后这些化合物的合成需要活化的糖类,例如udp-n-乙酰葡糖胺、udp-n-乙酰半乳糖胺、cmp-唾液酸、gdp-岩藻糖……,这些非常昂贵且难以合成分子并且由于其生物合成所需的能量而最好由活的生长细胞产生。
人/母乳的寡糖组分具有抗炎和益生元效果和/或在治疗学中用作营养品、抗炎剂、益生元(prebiotic)或药物(15,24,68)。然而,仍然需要有效生产后面的高价值化合物的方法。
本发明描述了即使在高乳糖浓度下也不导致乳糖杀伤的乳糖转运蛋白的合成表达系统。因此,能够通过所述表达系统获得的突变生物体可用于生产上述生物产品。
附图说明
图1:乳糖对大肠杆菌野生型菌株的影响。在箭头处,将乳糖添加到培养物之一中,并且该培养物的生长立即停止,而其他菌株在其他培养物中继续生长。
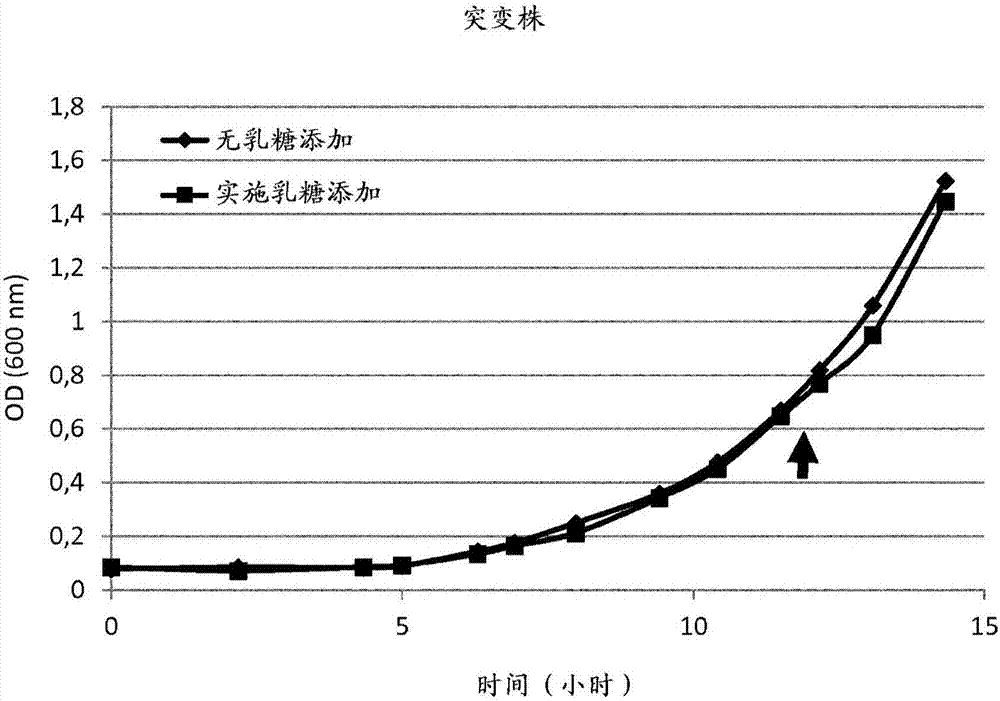
图2:乳糖对大肠杆菌乳糖转运蛋白突变株(mutantstrain)的影响,在所述突变株中通过合成的组成型启动子改变了乳糖转运蛋白表达。箭头指示将乳糖添加到培养物之一的培养基中的时刻。在这种情况下,未观察到对生长的影响。因此,可以以这种方式选择这些突变株。
图3:当引入到大肠杆菌突变株中时不引起乳糖杀伤的启动子、rbs和乳糖转运蛋白序列组合的实例(seqidn°1)。
图4:与lacz基因翻译偶联的乳糖通透酶基因的序列的实例(seqidn°2)。
图5:与cat基因翻译偶联的乳糖通透酶基因lacy的序列的实例(seqidn°3)。
图6:与aph1基因翻译偶联的马克斯克鲁维酵母(k.maxianus)乳糖通透酶基因的序列的实例(seqidn°4)。
图7:与适配体偶联的乳糖通透酶基因lacy的序列的实例(seqidn°5),所述适配体结合(z)-4-(3,5-二氟-4-羟基亚苄基)-1,2-二甲基-1h-咪唑-5(4h)-酮并允许检测乳糖通透酶表达。
图8:包含参考质粒psc101而不含与氯霉素抗性基因翻译偶联的乳糖通透酶的参考菌株以及具有与氯霉素抗性基因翻译偶联的乳糖通透酶的突变株的氯霉素抗性。x轴显示测试的不同氯霉素浓度,y轴显示培养物在孵育48小时后的光密度。参考菌株在比突变株更低的氯霉素筛选下显示出生长阻滞,这证明,可以通过与抗生素抗性基因的翻译偶联来筛选乳糖通透酶的表达。因此,可以以这种方式从不表达和表达乳糖通透酶的生物体混合物选择表达乳糖通透酶的生物体。
图9:包含参考质粒psc101而不含与氯霉素抗性基因翻译偶联的乳糖通透酶的参考菌株以及具有与氯霉素抗性基因翻译偶联的乳糖通透酶的突变株的氯霉素抗性。x轴显示测试的不同氯霉素浓度,y轴显示培养物在孵育92小时后的光密度。参考菌株在比突变株更低的氯霉素筛选下显示出生长阻滞,这证明,可以通过与抗生素抗性基因的翻译偶联来筛选乳糖通透酶的表达。因此,可以以这种方式从不表达和表达乳糖通透酶的生物体混合物选择表达乳糖通透酶的生物体。
图10:pcxp14-ft_幽门螺杆菌(h.pylori)(seqidn°6)。
图11:乳糖对酵母野生型菌株(乳酸马克斯克鲁维酵母(kluyveromycesmarxianuslactis))的影响。在箭头处,将乳糖添加到培养物之一中,并且该培养物的生长立即停止,而其他菌株在其他培养物中继续生长。
图12:乳糖对酵母乳糖转运蛋白突变株(酿酒酵母(saccharomycescerevisiae))的影响,在所述突变株中通过合成的组成型启动子改变乳糖转运蛋白表达。箭头指示将乳糖添加到培养物之一中的培养基中的时刻。在这种情况下,未观察到对生长的影响。因此,可以以这种方式选择这些突变株。
图13:当引入到酵母突变株中时不引起乳糖杀伤的启动子(p1)kozak、马克斯克鲁维酵母乳糖通透酶编码序列和终止子组合的实例(seqidn°7)。
图14:当引入到酵母突变株中时不引起乳糖杀伤的启动子(p2)kozak、马克斯克鲁维酵母乳糖通透酶编码序列和终止子组合的实例(seqidn°8)。
图15:启动子、kozak、马克斯克鲁维酵母β-半乳糖苷酶编码序列和终止子组合的实例(seqidn°9)。
图16:hr1rdna(seqidn°10)。
图17:hr2rdna(seqidn°11)。
图18:所选择的来源于如实施例中所述的乳糖杀伤筛选方法的序列的实例(seqidn°12至57)。
图19:乳糖通透酶表达盒相对于野生型的相对生长速率。误差条是至少3次重复测量的标准偏差。与x轴上的序列号相关的序列示于图18中。
图20:实施例16中对乳糖杀伤进行测试的laciq_lacy表达盒的序列(seqidn°58)。
图21:乳糖对大肠杆菌野生型菌株和placiq_lacy突变株的影响。两种菌株均在指数期中期在添加和不添加乳糖的情况下生长。箭头表示添加乳糖的时刻。两种菌株的生长由于添加乳糖而严重受损。
技术实现要素:
本发明涉及产生微生物的方法,所述微生物当在乳糖与其他碳源组合的环境中生长时抵抗乳糖杀伤现象,其中所述方法包括:
a.使微生物内乳糖转运蛋白的表达突变,其中所述突变导致表达的乳糖转运蛋白,
b.使所述突变微生物在包含非乳糖的碳源的培养基上生长,
c.在所述突变微生物的生长期间向所述培养基中添加乳糖,和
d.选择在所述包含乳糖的培养基上生长时抵抗乳糖杀伤现象的微生物。
更具体地,本发明涉及生产微生物的方法,所述微生物当在乳糖与其他碳源组合的环境中生长时抵抗乳糖杀伤现象,其中所述方法包括:
a.使微生物内乳糖转运蛋白的表达突变,其中所述突变导致所述乳糖转运蛋白表达,
b.使所述突变微生物在包含非乳糖的碳源的培养基上生长,
c.在所述突变微生物的生长期间向所述培养基中添加乳糖,和
d.选择这样的微生物,其在所述包含乳糖的培养基上生长时抵抗乳糖杀伤现象,并且保留用所述乳糖转运蛋白的野生型表达盒获得的乳糖通量的至少50%。
本发明还涉及如上所述方法,其中步骤a)通过以下来进行,在内源或外源乳糖转运蛋白基因前引入异源启动子,和/或对编码序列前的含有核糖体结合或kozak序列的非翻译区进行突变,和/或对内源乳糖转运蛋白基因的密码子使用进行修饰。
本发明还涉及所述方法,其中所述在内源或外源乳糖转运蛋白基因前引入异源启动子如下进行:a)从基因组中缺失内源乳糖转运蛋白并将其重新引入在所述微生物基因组内的另一位置处,或者b)在内源乳糖转运蛋白前引入异源启动子,或者c)敲除内源乳糖启动子并在所述微生物的基因组中的相同位置处引入异源启动子。
本发明还涉及上述方法,其中通过与报道基因翻译偶联和/或通过适配体偶联来检测步骤b)中所述乳糖通透酶的表达。
本发明涉及上述方法,其中通过如由seqidn°2、3、4和/或5给出的遗传构建体来检测所述经表达的乳糖转运蛋白。
本发明还涉及如上所述方法,其中所述乳糖转运蛋白是乳糖通透酶。
本发明还涉及如上所述方法,其中所述微生物是细菌、酵母或真菌。
本发明还涉及导致乳糖转运蛋白表达的,当含有这样的序列的微生物在乳糖与其他碳源组合的环境中生长时不导致乳糖杀伤表型的,并且能够通过上述方法获得的启动子序列、编码序列前的含有核糖体结合序列或kozak序列的非翻译区和/或乳糖通透酶序列。
本发明还涉及当在乳糖与其他碳源组合的环境中生长时抵抗乳糖杀伤现象并且能够通过上述方法获得的微生物。
更具体地,本发明涉及如上所述微生物,并且所述微生物在如由seqidn°1、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56或57给出的乳糖转运蛋白基因前具有异源序列。
本发明还涉及抵抗乳糖杀伤现象的微生物,其中使得编码来自乳糖和/或半乳糖降解途径之酶的基因功能降低或无功能。
本发明还涉及使用如上所述微生物来生产基于乳糖的特殊产品,例如特殊的碳水化合物、糖脂和半乳糖基化化合物。
本发明还涉及使用如上所述微生物,其中所述特殊碳水化合物是2-岩藻糖基乳糖、或2’-岩藻糖基乳糖、或3-岩藻糖基乳糖、或2’,3-二岩藻糖基乳糖、或乳糖基n三糖、或乳糖基-n-四糖、或乳糖基-nn-四糖、或3’唾液酸基乳糖(sialyllactose)或6’唾液酸基乳糖。
具体实施方式
本发明描述了通过在生物体中经由基因工程改变乳糖转运蛋白的表达来避免乳糖杀伤的新方法,得到表达突变体乳糖转运蛋白的生物体。为此,由不导致乳糖杀伤表型的异源启动子表达外源和/或内源乳糖转运蛋白基因,和/或对乳糖转运蛋白的密码子使用进行修饰以产生不导致乳糖杀伤表型的乳糖转运蛋白的表达改变。为此,乳糖转运蛋白的天然表达控制必须以这样的不发生乳糖杀伤的方式被除去和/或替代。例如,从基因组中缺失天然存在的乳糖转运蛋白表达盒并在另一位置重新引入,和/或,将异源启动子在其原始位置处引入到乳糖转运蛋白前,和/或,首先敲除乳糖转运蛋白并在基因组中相同和/或另一位置处用异源启动子重新引入,和/或,将乳糖转运蛋白引入到通过异源启动子表达的操纵子中。
因此,本发明涉及生产微生物的方法,所述微生物在乳糖与其他碳源组合的环境中生长时抵抗乳糖杀伤现象,其中所述方法包括:1)使微生物内乳糖转运蛋白的表达突变,其中所述突变导致所述乳糖转运蛋白表达,2)使所述突变的微生物在包含非乳糖的碳源的培养基上生长,3)在所述突变的微生物的生长期间向所述培养基中添加乳糖,以及4)选择这样的微生物,其在所述包含乳糖的培养基上生长时抵抗乳糖杀伤现象,并且保留用所述乳糖转运蛋白的野生型表达盒获得的乳糖通量的至少50%。
术语“乳糖杀伤”是指在乳糖或半乳糖苷与其他碳源组合的环境中生长的生物体的生长阻滞(growthretardation)或生长停滞现象。这些碳源是但不限于甘油、麦芽糖、葡萄糖、果糖、蔗糖、岩藻糖、甘露糖、唾液酸、淀粉、纤维素、多元醇(例如甘露醇、木糖醇、山梨糖醇)、有机酸(乳酸盐/酯、琥珀酸盐/酯、乙酸盐/酯……)和/或戊糖(木糖、阿拉伯糖……)。
本发明描述了鉴定不导致乳糖杀伤的乳糖通透酶表达系统的方法。该方法包括突变株的生长分析,其中在指数期中期向所述突变株添加乳糖。该方法可以以高通量方式在微量滴定板中或用细胞分选仪进行,使得能够筛选多个启动子、核糖体结合位点、密码子使用以及其他可影响突变微生物中乳糖通透酶的表达的因子。本发明描述了通过翻译偶联和/或适配体偶联来检测乳糖通透酶表达以及通过报道基因选择导致表达的序列的方法,所述报道基因例如但不限于抗生素抗性基因(例如但不限于氯霉素、遗传霉素g418)、荧光蛋白、水解酶(例如但不限于半乳糖苷酶、木聚糖酶)和或适配体序列。基于报道基因通过在具有抗生素的培养基中生长、比色测定(例如x-gal)和/或通过分选出荧光细胞的荧光激活细胞分选仪和/或适配体测定进一步选择导致乳糖通透酶表达的序列,例如但不限于基于(z)-4-(3,5-二氟-4-羟基亚苄基)-1,2-二甲基-1h-咪唑-5(4h)-酮(37,64)。本发明还描述了筛选不经历乳糖杀伤的表达乳糖转运蛋白的突变生物体的过程。此外,本发明描述了如何能通过乳糖通透酶基因的启动子文库、rbs或kozak序列文库、转录终止子文库和/或密码子使用变体来创建乳糖通透酶表达盒的文库。这些文库通过例如但不限于gibson组装、goldengate组装、cliva组装、lcr或限制性连接的方法进一步创建(25,36,50,79)。
术语“其保留用所述乳糖转运蛋白的野生型表达盒获得的乳糖通量的至少50%”涉及以下事实:本发明的突变微生物应当仍然能够保留当使用所述乳糖转运蛋白的野生型表达盒时获得的乳糖通量的至少50%(即,50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、99%),尽管它们抵抗乳糖杀伤。
术语“表达盒”涉及其中存在启动子序列、非翻译区序列(包含核糖体结合序列或kozak序列)、编码序列(例如乳糖通透酶基因序列)和任选的转录终止子(18)的任何序列。
术语“乳糖转运蛋白”是指在微生物中表达的能够转运(运输)乳糖穿过细胞质膜的任何蛋白质。这样的蛋白质是例如乳糖通透酶(来自超家族mfs或主要易化子超家族(majorfacilitatorsuperfamily)的转运蛋白)。
术语“异源启动子”是指不天然存在于编码序列前的任何启动子。“启动子”是位于转录起始位点和编码序列前的非翻译区之前的全部rna聚合酶结合序列。因此,“异源启动子”序列是:1)含有至少1个(即,1、2、3、4……)突变的天然存在的启动子序列的变体;和/或2)来自表达突变乳糖转运蛋白的微生物的天然启动子,所述启动子不天然存在于所述转运蛋白的编码序列之前;和/或3)不天然存在于表达乳糖转运蛋白的微生物中的序列;和/或4)为计算机设计的启动子的人工启动子。这些启动子可来源于文库,例如但不限于由alper等(2005)、hammer等(2006)、demey等(2007)、coussement等(2014)或mutalik等(2013)(3,25,29,41,60)(66)或blount等(2012)所述的文库,和/或hxt7p,启动子例如但不限于adh1p、tef1p、tef2p、gpdp、pdc1p、fba1p、pgk1p、pgi1p、tdh2p、pyk1p、eno2p、gpm1p、tpi1p(13)或者例如由rhodius等(2012)所述设计的启动子。术语“人工启动子”还指具有为天然(自体)启动子序列与部分不同(自体或异源)启动子序列之组合的dna序列的启动子。这样的“人工启动子”的序列可以存在于数据库(例如partsregistry.org(19))中。异源启动子导致组成型表达或通过转录因子的受调节表达。
术语“组成型表达”被定义为在某些生长条件下不受除rna聚合酶亚基(例如,细菌σ因子)以外的转录因子调节的表达。这样的转录因子的非限制性实例是大肠杆菌中的crp、laci、arca、cra、iclr……,或酿酒酵母中的aft2p、crz1p、skn7……,或枯草芽孢杆菌(b.subtilis)中的deor、gntr、fur……。这些转录因子在特定序列上结合并且可以阻断或增强在某些生长条件下的表达。rna聚合酶结合特定序列以启动转录,例如通过原核宿主中的σ因子。
术语“受调节表达”被定义为在某些生长条件下还受rna聚合酶亚基(例如,细菌σ因子)以外的转录因子调节的表达。这样的转录因子的实例如上所述。
术语在编码序列前的包含核糖体结合位点或kozak序列的“非翻译区”涉及rna聚合酶结合序列与编码序列之间的序列。这种非翻译区也是天然存在于编码序列前的序列,和/或天然存在于表达乳糖转运蛋白的微生物中的序列,和/或来源于其他生物体(原核生物或真核生物)的序列,和/或人工设计的序列,其涉及具有已知或可测量的翻译速率的非天然或计算机设计的核糖体结合位点,这些序列可来源于如由mutalik等(2013)(60)所述的文库或者通过例如由salis等(2009)(67)所述的算法设计或者可以存在于数据库(例如partsregistry.org(19))中。
术语“对密码子使用进行修饰”涉及将dna编码序列中使用的密码子改变为生物体更频繁使用或生物体很少使用的密码子。在数据库如密码子使用数据库(61)中定义了密码子使用并且通过密码子使用设计算法(73)优化密码子使用。
术语“翻译偶联”是指目的基因和报道基因(例如绿色荧光蛋白、抗生素抗性基因、毒性基因)的偶联表达(49,53,58,63)。术语“翻译传感器”是指作为基因表达和翻译的指示的任何机制,例如,如以下参考文献(17,72)中所述的荧光标签和分离标签。
术语“适配体偶联”是指将适配体序列引入到可通过荧光团检测的乳糖转运蛋白的信使rna中,所述荧光团例如但不限于(z)-4-(3,5-二氟-4-羟基亚苄基)-1,2-二甲基-1h-咪唑-5(4h)-酮(37,64)。
术语“生长分析”是指生物体的生长曲线的分析。在生长环境中的生长培养基中培养该生物体。术语生长环境涉及所有环境参数,例如ph、温度、溶解氧。通过ph缓冲液或通过ph控制设置ph并且例如但不限于ph4、4.5、5、5.5、6、6.5、7、7.5或8。将温度设置为例如但不限于25℃、28℃、30℃、32℃、34℃、37℃、40℃、42℃、45℃。溶解氧为厌氧、微需氧(溶解氧低于1.5mg/l)或需氧条件。
术语“培养基”或“生长培养基”涉及包含生物体生长所必需的基质的任何溶液。这些基质是但不限于:氮源,例如铵盐、硝酸盐;酵母提取物;蛋白胨(pepton);酪蛋白氨基酸(casamino)和/或氨基酸;磷源,例如但不限于磷酸盐;硫源,例如但不限于硫酸盐;元素,例如但不限于铜、钴、铁、硒、碘、钼酸盐、镁、钙、钾、钠、锌、镍、锰;和/或硼酸;和/或维生素,例如但不限于硫胺素、泛酸和/或烟酸;和/或碳源,例如但不限于甘油、麦芽糖、葡萄糖、果糖、蔗糖、岩藻糖、甘露糖、唾液酸、淀粉、纤维素、多元醇(例如甘露醇、木糖醇、山梨糖醇)、有机酸(乳酸盐、琥珀酸盐、乙酸盐……)和/或戊糖(木糖、阿拉伯糖……)。
如上所述术语“生物体”或“细胞”是指选自细菌、酵母或真菌的微生物,或者指植物或动物细胞。前述细菌优选地属于变形菌(proteobacteria)门或厚壁菌(firmicutes)门或蓝细菌(cyanobactria)门或者异常球菌-栖热菌(deinococcus-thermus)门。属于变形菌门的前述细菌优选地属于肠杆菌科(enterobacteriaceae),优选地属于大肠杆菌(escherichiacoli)种。前述细菌优选地涉及属于大肠杆菌种的任何菌株,例如但不限于大肠杆菌b、大肠杆菌c、大肠杆菌w、大肠杆菌k12、大肠杆菌nissle。更具体地,前述术语涉及培养的大肠杆菌菌株-称为大肠杆菌k12菌株-其非常适合于实验室环境,并且与野生型菌株不同,其已丧失了在肠中繁殖的能力。大肠杆菌k12菌株的公知实例是k12野生型、w3110、mg1655、m182、mc1000、mc1060、mc1061、mc4100、jm101、nzn111和aa200。因此,本发明特别涉及如上所述突变和/或转化的大肠杆菌菌株,其中所述大肠杆菌菌株是k12菌株。更具体地,本发明涉及如上所述突变和/或转化的大肠杆菌菌株,其中所述k12菌株是大肠杆菌mg1655。属于厚壁菌门的前述细菌优选地属于芽孢杆菌属(bacilli),优选地来自芽孢杆菌种。前述酵母优选地属于子囊菌门(ascomycota)或担子菌门(basidiomycota)或半知菌门(deuteromycota)或接合菌门(zygomycete)。前述酵母优选地属于酵母属(saccharomyces)、毕赤酵母属(pichia)、汉逊酵母属(hansunella)、克鲁维酵母属(kluyveromyces)、耶氏酵母属(yarrowia)或斯塔摩酵母属(starmerella)。前述真菌优选地属于根霉属(rhizopus)、盘基网柄菌属(dictyostelium)或曲霉属(aspergillus)。
本发明描述了能够摄取乳糖而不经历乳糖杀伤并且能够将乳糖或其半乳糖部分转化为特殊产品的生物体。更具体地,所述特殊产品或生物产品是特殊的碳水化合物;糖脂,例如但不限于半乳糖脂和/或乳糖脂;和/或半乳糖基化化合物,例如半乳糖基脂、半乳糖神经酰胺和/或半乳糖基化糖苷配基。
本发明描述了能够摄取乳糖而不经历乳糖杀伤并将所述乳糖转化为人乳寡糖的生物体,所述人乳寡糖例如但不限于3-岩藻糖基乳糖、2’-岩藻糖基乳糖、6-岩藻糖基乳糖、2’,3-二岩藻糖基乳糖、2’,2-二岩藻糖基乳糖、3,4-二岩藻糖基乳糖、6’-唾液酸基乳糖、3’-唾液酸基乳糖、3,6-二唾液酸基乳糖、6,6’-二唾液酸基乳糖、3,6-二唾液酸基乳糖基-n-四糖、乳糖基二岩藻糖基丁糖(lactodifucotetraose)、乳糖基-n-四糖、乳糖基-n-新四糖、乳糖基-n-岩藻糖基五糖ii、乳糖基-n-岩藻糖基五糖i、乳糖-n-岩藻糖基五糖iii、唾液酸基乳糖基-n-四糖c、唾液酸基乳糖基-n-四糖b、唾液酸基乳糖基-n-四糖a、乳糖基-n-二岩藻基己糖i、乳糖基-n-二岩藻基己糖ii、乳糖基-n-己糖、乳糖基-n-新己糖、对乳糖基-n-己糖、单岩藻糖基单唾液酸基乳糖基-n-四糖c、单岩藻糖基对乳糖基-n-己糖、单岩藻糖基乳糖基-n-六糖iii、异构岩藻糖基化乳糖基-n-己糖iii、异构岩藻糖基化乳糖基-n-己糖i、唾液酸乳糖基-n-己糖、唾液酸乳糖基-n-新己糖ii、二岩藻糖基对乳糖基-n-己糖、二岩藻糖基乳糖基-n-己糖、二岩藻糖基乳糖基-n-己糖a、二岩藻糖基乳糖基-n-己糖c、半乳糖基化壳聚糖、岩藻糖基化寡糖和/或唾液酸化寡糖……。
本发明还描述了不经历乳糖杀伤并且合成核苷酸糖的生物体,所述核苷酸糖例如但不限于gdp-l-岩藻糖、gdp-甘露糖、gdp-葡萄糖、cmp-唾液酸、udp-葡萄糖、udp-半乳糖、udp-n-乙酰葡糖胺、udp-n-乙酰甘露糖胺、udp-n-乙酰半乳糖胺、udp-葡糖醛酸、udp-半乳糖醛酸、udp-木糖、udp-阿拉伯糖和/或dtdp-鼠李糖。术语“唾液酸”是例如但不限于由varki(1992)(71)定义的神经氨酸、n-乙酰神经氨酸或n-羟乙酰神经氨酸(glycoylneuramicacid)的化合物的组名。细胞内gdp-岩藻糖浓度或库通过上调从头途径和/或补救途径而增强。从头途径由gdp-4-酮-6-脱氧甘露糖-3,5-差向异构酶-4-还原酶或gdp-岩藻糖合酶、gdp-甘露糖-4,6-脱水酶、gdp-d-甘露糖焦磷酸化酶、磷酸甘露糖异构酶和/或磷酸甘露糖变位酶组成,其表达通过如下单独增强:遗传元件,例如但不限于单顺反子或多顺反子(操纵子结构)中的启动子和/或核糖体结合位点和/或改变的密码子使用;和/或通过肠杆菌科(enterobacteriaceae)中的调节子arcaiclr和/或通过肠杆菌科中的转录调节子rcsa或细菌中的同源基因,或酿酒酵母中的xbp1、spt20、sfp1、rpd3、rap1、gcr1、gcn5、cst6、abf1、hsf1、reb1、cad1、sin4、ash1、ixr1、met32、pho2、rgr1、spt7、swi4,或酵母或真菌中的同源基因。补救途径由l-岩藻糖激酶和/或gdp-l-岩藻糖焦磷酸化酶组成。gdp-甘露糖库通过上调编码gdp-d-甘露糖焦磷酸化酶、磷酸甘露糖异构酶和/或磷酸甘露糖变位酶的基因和/或编码甘露糖激酶和gdp-d-甘露糖焦磷酸化酶的基因而增强。udp-半乳糖库通过上调编码如下的基因而增强:半乳糖激酶和/或半乳糖1-磷酸尿苷酰转移酶;和/或udp-半乳糖-4-差向异构酶;和/或udp-半乳糖/葡萄糖焦磷酸化酶;和/或乳糖合酶;和/或乳糖磷酸化酶;和/或蔗糖磷酸化酶。udp-葡萄糖库通过上调编码如下的基因而增强:葡糖激酶;和/或udp-葡萄糖焦磷酸化酶;和/或蔗糖磷酸化酶;和/或磷酸葡糖变位酶;和/或蔗糖合酶。cmp-唾液酸库通过上调编码如下的基因而增强:l-谷氨酰胺:d-果糖-6-磷酸转氨酶;和/或磷酸葡糖胺变位酶;和/或葡糖胺-1-磷酸乙酰转移酶;和/或n-乙酰葡糖胺-1-磷酸尿苷酰转移酶;和/或udp-n-乙酰葡糖胺-2-差向异构酶;和/或n-乙酰神经氨酸合酶(n-acetylneuraminatesynthase);和/或胞苷5’-单磷酸n-乙酰神经氨酸合成酶。udp-n-乙酰葡糖胺库通过上调编码如下的基因而增强:l-谷氨酰胺:d-果糖-6-磷酸转氨酶;和/或磷酸葡糖胺变位酶;和/或葡糖胺-1-磷酸乙酰转移酶;和/或n-乙酰葡糖胺-1-磷酸尿苷酰转移酶;和/或葡糖胺-6-磷酸n-乙酰转移酶;和/或磷酸乙酰葡糖胺变位酶;和/或udp-n-乙酰葡糖胺焦磷酸化酶。udp-n-乙酰甘露糖胺库通过上调编码udp-n-乙酰葡糖胺库增强和/或udp-n-乙酰葡糖胺2-差向异构酶的基因而增强。udp-n-乙酰半乳糖胺库通过上调编码udp-n-乙酰葡糖胺库增强和/或udp-n-乙酰葡糖胺c4-差向异构酶的基因而增强。udp-葡糖醛酸库通过上调编码udp-葡萄糖库增强和/或udp-葡萄糖脱氢酶的基因而增强。udp-木糖库通过上调编码udp-葡糖醛酸库(glucuronicpool)增强和/或udp-d-木糖合酶的基因而增强。udp-半乳糖醛酸库通过上调编码udp-葡糖醛酸库增强和/或udp-d-葡糖醛酸4-差向异构酶的基因而增强。udp-阿拉伯糖库通过上调编码如下的基因而增强:udp-葡糖醛酸库增强和/或udp-d-木糖4-差向异构酶和/或阿拉伯糖激酶和/或udp-l-阿拉伯糖焦磷酸化酶。dtdp-鼠李糖库通过上调编码如下的基因而增强:dtdp-葡萄糖焦磷酸化酶;和/或dtdp-葡萄糖4,6-脱水酶;和/或dtdp-4-脱氢鼠李糖3,5-差向异构酶;和/或dtdp-4-脱氢鼠李糖还原酶;和/或葡萄糖-1-磷酸胸苷酰转移酶(thymidylyltransferase);和/或核苷酸鼠李糖合酶。
术语“库”还涉及天然存在于野生型生物体中的代谢物的浓度,例如,核苷酸糖库的浓度。术语“增强库”涉及所述代谢物库的比野生型生物体之代谢物库高的显著提高的浓度。
术语“上调基因”涉及导致基因表达和/或产物(如果所述基因有的话)活性增强的每种基因修饰。所述基因修饰是启动子、非翻译区、核糖体结合位点、编码序列、基因位置、内含子/外显子结构和/或转录终止子中的修饰,其导致所述提高的表达和/或活性。
另外,本发明描述了可通过糖基转移酶将这些核苷酸糖转移到单糖、二糖或寡糖上的遗传修饰的生物体,所述单糖、二糖或寡糖例如但不限于:半乳糖、乳糖、乳糖基n二糖、乳糖基n三糖、乳糖基n四糖、乳糖基-n-新四糖、globotriose、2’-岩藻糖基乳糖、3-岩藻糖基乳糖、3-唾液酸基乳糖、6-唾液酸基乳糖、人乳寡糖、heparosan、壳聚糖、结瘤因子、糖脂和/或糖苷配基……。
本发明还描述了不经历乳糖杀伤并且可以用酶进一步修饰所述乳糖的生物体,所述酶例如但不限于:碳水化合物水解酶、糖碳水化合物转移酶、碳水化合物合酶、乙酰化酶、酰基转移酶、碳水化合物磷酸酶、多糖裂解酶、激酶、丙酮酸酶(pyruvylase)和/或磺基转移酶。
本发明还描述了不经历乳糖杀伤且通过使乳糖水解酶基因具有较少功能或无功能而不再降解乳糖的生物体。
术语“使得基因具有较少功能或无功能”是指技术人员公知的技术,例如使用sirna、rnai、mirna、asrna、使基因产生突变、敲除基因、转座子诱变、crispr/cas等,其用于以这样的方式改变基因:它们不太可能(即,与功能野生型基因相比统计学上显著“不太可能”)或完全不能(例如敲除基因)产生功能最终产物。因此,术语“(基因)敲除”是指使得基因无功能。术语“缺失基因”或“基因缺失”也指使得基因无功能(2,4-9,22,27,30,43,45,46,51,65,74)。
本发明描述了其中引入/敲入/上调基因以产生如上所述的生物产品的生物体。这些基因存在于基因数据库中,例如但不限于基因库或蛋白质数据库(例如但不限于uniprot),或酶数据库(例如但不限于brenda酶数据库(16,23,38)),并且针对生物产品的途径存在于在数据库中,例如但不限于kegg、biocyc、metacyc(20,44,47)。针对上述某一生物产品的途径可通过本领域中所述的若干种数学工具(35,54,57,76)确定。
实施例
1.材料和方法大肠杆菌
菌株和质粒
大肠杆菌mg1655[λ-,f-,rph-1]和jm109获自荷兰细菌培养物保藏中心(netherlandsculturecollectionofbacteria,nccb)。使用datsenko&wanner的方法(27)产生所有突变株。
培养基
luria肉汤(luriabroth,lb)培养基由1%胰蛋白胨蛋白胨(difco,erembodegem,belgium)、0.5%酵母提取物(difco)和0.5%氯化钠(vwr,leuven,belgium)组成。摇瓶培养基包含2g/lnh4cl、5g/l(nh4)2so4、2.993g/lkh2po4、7.315g/lk2hpo4、8.372g/lmops、0.5g/lnacl、0.5g/lmgso4·7h2o、15g/l甘油(除非另有说明)、1ml/l维生素溶液、100μl/l钼酸盐溶液和1ml/l硒溶液。用1mkoh将培养基设置为ph为7。
维生素溶液由3.6g/lfecl2·4h2o、5g/lcacl2·2h2o、1.3g/lmncl2·2h2o、0.38g/lcucl2·2h2o、0.5g/lcocl2·6h2o、0.94g/lzncl2、0.0311g/lh3bo4、0.4g/lna2edta·2h2o和1.01g/l硫胺·hcl组成。钼酸盐溶液包含0.967g/lna2moo4·2h2o。硒溶液包含42g/lseo2。
用于发酵的基本培养基包含6.75g/lnh4cl、1.25g/l(nh4)2so4、1.15g/lkh2po4、0.5g/lnacl、0.5g/lmgso4·7h2o、30g/l乳糖和20g/l蔗糖(或不同浓度,如果另有说明),具有与上述相同组成的1ml/l维生素溶液、100μl/l钼酸盐溶液和1ml/l硒溶液。
培养条件
将来自lb-板上单菌落的预培养物在5mllb培养基中在37℃下在轨道摇床(orbitalshaker)上以200rpm孵育8小时。从该培养物中,将2ml转移到500ml摇瓶中的100ml基本培养基中,并在37℃下在轨道摇床上以200rpm孵育16小时。将4%接种物用于具有1.5l或4l工作体积的2或5lbiostatbplus培养容器(sartoriusstedimbiotech,melsungen,germany)中。培养条件为:37℃,以800rpm搅拌,并且气体流速为1.5l/分钟。通过用空气鼓泡来维持需氧条件。用0.5mh2so4和35%m氨溶液将ph维持在7。通过排气冷却器(frigomix1000,sartoriusstedimbiotech,melsungen,germany)使废气冷却至4℃。当发酵期间发泡产生时添加硅酮消泡剂(bdh331512k,vwrintltd.,poole,england)的10%溶液(约10μl)。用el3020尾气分析仪(abbautomationgmbh,60488frankfurtammain,germany)测量尾气。
用sartoriusmfcs/winv3.0系统(sartoriusstedimbiotech,melsungen,germany)记录所有数据。
抽样方法
生物反应器在其内部包括连接至反应器端口的收获管(bdspinalneedle,1.2x152mm(bdmedicalsystems,franklinlakes,nj-usa)),其在外部连接至masterflex-14管(cole-parmer,antwerpen,belgium),随后是具有隔膜的收获端口用于取样。该收获端口的另一侧用masterflex-16管连接回反应容器。该系统被称为快速取样回路。在取样期间,在取样回路中循环泵送反应器肉汤(reactorbroth)。据估计,在150ml/分钟的流速下,反应器肉汤需要0.04秒到达收获端口,并且需要3.2秒重新进入反应器。在50%的po2水平下,在37℃的液体中存在约3mg/l的氧。po2水平决不应低于20%以避免微需氧条件。因此,在运输通过收获回路的过程中可以消耗1.8mg/l的氧。假设摄氧速率为0.4g氧/g生物质/小时(在μmax存在的最大摄氧速率),这得到5g/l生物质,2g/l/小时或0.56mg/l/秒的摄氧速率乘以3.2秒(在回路中的停留时间)得到1.8mg/l耗氧量。
为了停止取样期间细胞的代谢,将反应器肉汤抽吸通过填充有在-20℃下预冷却的62g不锈钢珠的注射器中的收获端口,即刻将5ml肉汤冷却至4℃。取样后立即冷离心(15000g,5分钟,4℃)。在分批实验期间,使用快速取样回路和冷不锈钢珠取样方法取得用于od600nm测量的样品。
分析方法
通过在600nm处测量光密度(uvikom922分光光度计,brs,brussel,belgium)来频繁监测培养物的细胞密度。通过将预干燥和称重的falcon中的20g反应器肉汤离心(15分钟,5000g,gsa转子,sorvallrc-5b,goffinmeyvis,kapellen,belgium)来获得细胞干重。将沉淀随后用20ml生理溶液(9g/lnacl)洗涤一次并在70℃下干燥至恒重。为了能够将od600nm测量值转换为生物量浓度,作od600nm对生物量浓度的相关曲线。使用在65℃下加热的配备有1cm预置柱的aminexhpx-87h柱(bio-rad,eke,belgium),使用5mmh2so4(0.6ml/分钟)作为流动相,在varianprostarhplc系统(varian,sint-katelijne-waver,belgium)上测定葡萄糖和有机酸的浓度。使用双波uv-vis(210nm和265nm)检测器(varianprostar325)和示差折光率检测器(mercklachroml-7490,merck,leuven,belgium)进行峰检测。通过将在265nm和210nm两处的峰吸收相除,可以鉴定峰。除法得到恒定值,其对于某一化合物而言是典型的(beer-lambert公式)。
葡萄糖、果糖、蔗糖、寡糖和葡萄糖-1-磷酸通过具有hypercarb柱的hplc测量,并用msms检测器检测(antonio等,2007;nielsen等,2006)。
遗传方法
用于突变体构建的方法描述如下。
将质粒保持在宿主大肠杆菌dh5α(f-、
质粒。pkd46(red辅助质粒,氨苄青霉素抗性)、pkd3(包含frt侧翼的氯霉素抗性(cat)基因)、pkd4(包含frt侧翼的卡那霉素抗性(kan)基因)和pcp20(表达flp重组酶活性)质粒用于突变体构建。使用质粒pbluescript(fermentas,st.leon-rot,germany)来构建具有启动子文库或具有携带点突变的等位基因的pkd3和pkd4的衍生物。
突变。突变包括基因破坏(敲除,knock-out,ko)。使用datsenko和wanner的概念(27)将其引入。
使携带red辅助质粒的转化体在具有氨苄青霉素(100mg/l)和l-阿拉伯糖(10mm)的10mllb培养基中在30℃下生长至od600nm为0.6。通过第一次用50ml冰冷的水以及第二次用1ml冰冷的水洗涤细胞将其制成电感受态(electrocompetent)。然后,将细胞重悬于50μl冰冷的水中。通过使用genepulsertm(biorad)(600ω,25μfd和250伏特),用50μl细胞和10-100ng线性双链dna产物进行电穿孔。
电穿孔后,将细胞添加到1mllb培养基中,在37℃下孵育1小时,并且最后铺在包含25mg/l氯霉素或50mg/l卡那霉素的lb-琼脂上以选择抗生素抗性转化体。所选择的突变体通过用修饰区上游和下游的引物的pcr进行验证,并在42℃下在lb-琼脂中生长以失去辅助质粒。测试突变体的氨苄青霉素敏感性。
线性双链dna。使用pkd3、pkd4及其衍生物作为模板通过pcr获得线性双链dna扩增子。所使用的引物具有与模板互补的序列的一部分以及与染色体dna上必须发生重组的一侧互补的另一部分。对于ko,同源区域被设计为目的基因的起始和终止密码子上游50-nt和下游50-nt。对于ki,必须考虑转录起点(+1)。pcr产物进行pcr纯化,用dpni消化,从琼脂糖凝胶再纯化,并悬浮于洗脱缓冲液(5mmtris,ph8.0)中。
消除抗生素抗性基因。用pcp20质粒转化所选择的突变体(氯霉素或卡那霉素抗性),所述质粒显示出温度敏感型复制和热诱导的flp合成的氨苄青霉素和氯霉素抗性质粒。在30℃下选择氨苄青霉素抗性转化体,之后在42℃下在lb中对一些进行菌落纯化,然后测试所有抗生素抗性和flp辅助质粒的丢失。用对照引物检查基因敲除和敲入(fw/rv-基因验证(fw/rv-gene-out))。
转化。使用hanahan(42)的简化过程或通过如上所述的电穿孔在cacl2感受态细胞中转化质粒。
2.材料和方法酵母
2.1.菌株
酿酒酵母by4742获自euroscarf培养物保藏中心(euroscarfculturecollection)。使用gietz的方法(40)通过同源重组或质粒转化产生所有突变株。乳酸马克斯克鲁维酵母获自工业生物技术和生物催化实验室(laboratoryofindustrialbiotechnologyandbiocatalysis)的培养物保藏中心。
2.2.培养基
使菌株在具有完全补充混合物的合成的成分明确的酵母培养基(syntheticdefinedyeastmediumwithcompletesupplementmixture,sdcsm)或没有csm(csmdrop-out)的合成的成分明确的酵母培养基(sdcsm-ura)上生长,所述培养基包含6.7g·l-1无氨基酸的酵母氮源(yeastnitrogenbase)(ynbw/oaa,difco)、20g·l-1琼脂(difco)(固体培养物)、22g·l-1葡萄糖一水合物或20g·l-1乳糖和0.79g·l-1csm或0.77g·l-1csm-ura(mpbiomedicals)。
2.3.培养条件
首先将酵母培养物接种在5ml的合适的培养基中,并在30℃和200rpm下孵育过夜。为了获得更大体积的培养物,将2%(或更高)的预培养物接种在50-200ml培养基中。将这些培养物再次在30℃和200rpm下孵育。
在96孔板或erlenmeyer计量器(erlenmeyerscale)中进行生长实验。为了获得作为生长和生产实验的起始材料的单菌落,将菌株铺板在选择性sdcsm板上并在30℃下孵育2-3天。然后挑选一个菌落并转移至5ml培养基中用于erlenmeyer研究或转移至1ml培养基中用于微量滴定板实验。
对于erlenmeyer实验,将预培养物在30℃和200rpm下孵育过夜,并将2%的这些预培养物添加到100ml培养基中以开始生长实验。
对于mtp生长实验,将菌落添加到150μl培养基中并在30℃下孵育24小时。孵育后,将2μl的mtp预培养物添加到包含mtp的150μl新鲜培养基中。用
2.4.取样方法
每两小时直至稳定期和在稳定期期间每几个小时取培养物的od(0.2ml)样品以及细胞和上清液级分(1ml)样品两者。首先将1ml样品离心(11),之后分离细胞沉淀和上清液并在-20℃下分别储存。储存上清液用于细胞外产物分析,而沉淀用于细胞内代谢物分析。
2.5.分析方法
通过在600nm下测量光密度(uvikom922分光光度计,brs,brussel,belgium)或通过用biochromanthoszenyth340微量滴定板读数器来频繁监测培养物的细胞密度。通过将预干燥和称重的falcon中的20g反应器肉汤离心(15分钟,5000g,gsa转子,sorvallrc-5b,goffinmeyvis,kapellen,belgium)来获得细胞干重。随后将沉淀用20ml生理溶液(9g/lnacl)洗涤一次并在70℃下干燥至恒重。为了能够将od600nm测量值转换为生物量浓度,制作od600nm对生物量浓度的相关曲线。使用在65℃下加热的配备有1cm预置柱的aminexhpx-87h柱(bio-rad,eke,belgium),使用5mmh2so4(0.6ml/分钟)作为流动相,在varianprostarhplc系统(varian,sint-katelijne-waver,belgium)上测定葡萄糖和有机酸的浓度。使用双波uv-vis(210nm和265nm)检测器(varianprostar325)和示差折光率检测器(mercklachroml-7490,merck,leuven,belgium)进行峰检测。通过将在265nm和210nm两处的峰吸收相除,可以鉴定峰。除法得到恒定值,其对于某一化合物而言是典型的(beer-lambert公式)。
葡萄糖、果糖、蔗糖、寡糖和葡萄糖-1-磷酸通过具有hypercarb柱的hplc测量,并用msms检测器检测(antonio等,2007;nielsen等,2006)。
2.6遗传方法
用于突变体构建的方法描述如下。
将质粒保持在宿主大肠杆菌dh5α(f-、
质粒。使用可在工业生物技术和生物催化实验室处获得的酵母表达质粒p2a_2μ_lac4使得酵母属(saccharomyces)能够在作为唯一c源的乳糖上生长。该质粒包含氨苄青霉素抗性基因和细菌复制起点,以允许在大肠杆菌中进行选择和维持。质粒还包含2μ酵母ori和ura3选择标志物用于在酵母中选择和维持。最后,质粒包含β-半乳糖苷酶表达盒(seqid9,图15)。
突变。突变包括使用p2a_2μ_lac4(如上所述)的质粒引入和使用双链线性dna(如上所述)的基因敲入(ki)(在rdna基因座处的ki)。在用质粒dna转化后,将转化体铺在sdcsm-ura上,或者在用双链线性dna转化后,将其铺在用乳糖作为唯一c源的sdcsm-ura上。通过用匹配p2a_2μ_lac4的引物的pcr验证具有所选质粒的突变体。所选择的基因组敲入突变体通过用修饰区上游和下游的引物的pcr验证并通过测序确认(在lgcgenomics(lgcgroup,germany)处进行)。
线性双链dna。使用质粒pjet_ki_p1_lac12_t@rdna或pjet_ki_p2_lac12_t@rdna通过pcr获得线性双链dna扩增子。这些质粒在pjet克隆载体(thermoscientific)的多克隆位点处分别在seqid7和seqid8的侧翼包含2500bp的同源区(hr1(seqid10,图16)和hr2(seqid11,图17))。所使用的引物与hr1(正向引物)的5’端和hr2的3,端(反向引物)同源。pcr产物在转化前进行pcr纯化。
转化。使用方法gietz(40)转化质粒和线性双链dna。
3.结果
实施例1:在agp基因座处构建大肠杆菌乳糖通透酶lacy敲入
首先,根据如上所述datsenko和wanner的方法构建菌株mg1655δlacy。为此,用pkd46转化mg1655,并由基础质粒pkd3和pkd4构建与lacy基因侧翼同源的线性dna。然后用合适的抗生素筛选成功的重组。为了确保在该菌株中不摄取乳糖,在仅含有乳糖作为碳源的基本培养基上培养菌株。在该实验期间未观察到生长,因此该细胞不再能够运输乳糖通过其膜。
为了进一步构建合成表达系统,将合成启动子和rbs与lacy基因(从idt和geneart订购)组合一起合成。该序列示于图3。还通过改编datsenko和wanner方法将该序列在agp基因座处引入到基因组中。简言之,首先用来自pkd3质粒的筛选盒组装乳糖通透酶构建体,得到新质粒pcx_lacy-kan。由该质粒,可以pcr扩增与agp基因组区同源的线性dna。然后可将所获得的线性dna转化到大肠杆菌mg1655δlacy(其中存在pkd46质粒)中。这导致乳糖转运蛋白表达盒重组到基因组中,得到乳糖通透酶表达生物体mg1655δlacyδagp::lacy合成。为了确保恢复在乳糖上的生长,使该菌株在如上所述的乳糖基本培养基上生长。这导致完全恢复了在乳糖上的生长,生长速率类似于野生型。
实施例2:乳糖对野生型大肠杆菌菌株和不经历乳糖杀伤的突变大肠杆菌菌株的影响
用野生型mg1655和mg1655δlacyδagp::lacy合成建立如材料和方法中所述的摇瓶实验。使这些菌株在甘油摇瓶培养基(15g/l甘油,如材料和方法中所述)中生长,并在指数期中期(约在od0.8处)添加乳糖(添加200g/l储液,导致最终浓度为10g/l)。由图1和图2可以看出,突变菌株不经历乳糖杀伤。
实施例3:使用翻译偶联或翻译传感器(translationalsensor)以确保乳糖转运蛋白表达
由于完全乳糖通透酶敲除菌株也不会经历乳糖杀伤并且目标是获得有功能的、有活性的、表达的乳糖通透酶,因此需要筛选以确保乳糖通透酶表达。为此,可以产生启动子、核糖体结合位点、kozak序列、密码子使用和转录终止子的序列变体。然而,这些序列变体可导致无效表达构建体,因此导致乳糖转运蛋白阴性突变株。因此,需要设计系统以检测乳糖转运蛋白的表达,优选地通过易于筛选的报道基因如lacz、荧光蛋白或抗生素抗性基因。
重新启动报道基因的翻译以检测乳糖转运蛋白表达的翻译偶联系统的构建
通过在目的基因的3’和报道基因的5’之间引入翻译重新启动区可以翻译偶联两个基因。翻译可以通过若干个密码子重新启动,例如aug、uug、gug或aua(63)。这样的构建体(其将乳糖通透酶基因和lacz基因翻译连接)的序列示于图4。为了产生该序列,用具有goldengate限制性位点(bsai,获自neb)的引物从大肠杆菌基因组中扩增lacy和lacz序列。允许翻译偶联的基因间区域可以从任何基因合成公司(例如idt或geneart)订购。然后通过如由engler等(2013)(36)所述的goldengate方法将所有部分与如图3所示的启动子和rbs序列(对于大肠杆菌)一起组装到puc54质粒中或与芽孢杆菌启动子和核糖体结合位点一起组装到在芽孢杆菌属中复制的pgk12中。
通过打开核糖体结合位点上的环来启动报道基因的翻译以检测乳糖转运蛋白表达的翻译偶联系统的构建
mendez-perez等(2012)(58)描述了通过翻译偶联筛选表达的第二种方法。该方法适用于用氯霉素报道基因筛选乳糖通透酶表达。在这种情况下,乳糖转运蛋白lacy通过his-标签和核糖体结合位点与氯霉素抗性基因偶联。该构建体的序列部分也在idt或geneart处订购并通过goldengate组装来组装。所得序列在图5中给出。
将酵母乳糖转运蛋白与报道基因偶联的翻译偶联系统的构建
尽管酵母不使用顺反子,但仍可能经由病毒内部核糖体进入位点(所谓的ires序列)(56)通过翻译偶联来筛选表达。这样的序列的一个实例是t2a序列(10),其允许顺反子中两个蛋白质的完全独立(这意味着不是蛋白质融合)但偶联的翻译。这意味着如果顺反子的最后一个蛋白质得到表达,则第一个蛋白质也被表达。
在酵母中,例如马克斯克鲁维酵母的乳糖通透酶基因可用于在细胞中运输乳糖。该基因可以与t2a序列偶联至aph1基因,编码对遗传霉素的抗性。该序列类似地如上所述构建。最终序列在图6中给出。
将适配体引入乳糖通透酶的信使rna的适配体偶联系统的构建
乳糖通透酶表达也可以在信使rna水平上检测。为此,在如图7所示的乳糖通透酶编码序列后克隆(z)-4-(3,5-二氟-4-羟基亚苄基)-1,2-二甲基-1h-咪唑-5(4h)-酮结合适配体。如实施例6所述进一步调节该构建体的表达。在细胞生长后,如由pothoulakis等(2013)(64)所述,将(z)-4-(3,5-二氟-4-羟基亚苄基)-1,2-二甲基-1h-咪唑-5(4h)-酮添加到培养基中,并且通过荧光激活细胞分选仪(fluorescence-activatedcellsorter,facs)选择表达乳糖通透酶的突变株。
实施例4:检测与氯霉素抗性基因翻译偶联的乳糖转运蛋白的表达
构建了两种菌株,在所述菌株中从基因组中敲除了乳糖通透酶。将包含卡那霉素抗性基因的psc101质粒转化到两种菌株中,区别在于质粒之一包含如实施例1和2中所述的组成型表达的乳糖通透酶,得到如实施例4和图5所述的参考菌株mg1655δlacypsc101_kan和lacy_cat翻译偶联菌株mg1655δlacypsc101_kan_lacy合成_cat。使两种菌株在如上所述的基本培养基中在不同的氯霉素浓度(在0mg/l和30mg/l之间)下生长。图8和9显示在生长48和92小时之后,与突变体lacy_cat翻译偶联菌株相比,参考菌株的生长在更低的氯霉素浓度下受到抑制,这使得这样的系统能够优异的筛选表达乳糖通透酶的遗传构建体。
实施例5:不经历乳糖杀伤的表达乳糖通透酶的突变体的筛选步骤
类似于实施例2,使两种氯霉素抗性菌株(一种不经历乳糖杀伤且与氯霉素翻译偶联的菌株以及一种具有氯霉素盒但具有乳糖通透酶的天然表达系统的菌株)的混合物生长。如实施例2所述,使两种菌株在培养基中生长,并如实施例2所示在指数期中期添加乳糖。不经历乳糖杀伤的突变株保持生长,而另一种菌株(即,乳糖杀伤敏感的)停止生长。在指数期结束时,将0.1ml该培养物接种在具有如上所述相似培养基的第二个摇瓶中。再次,在od0.8处,添加乳糖,阻止乳糖杀伤敏感菌株的生长并进一步富集不经历乳糖杀伤的突变株。在该步骤重复5次后,获得99%富集的不经历乳糖杀伤的突变株,然后将其容易地从培养物中分离出来。
实施例6:乳糖通透酶表达盒的突变体的产生
启动子、核糖体结合位点、kozak序列、转录终止子的序列变体和乳糖通透酶基因变体(具有不同的密码子使用)在基因合成供应商(例如idt、geneart、genscript、gen9……)处订购。细菌启动子文库的设计基于细菌启动子的共有序列,在转录起始的-10bp和-35bp处具有两个保守区。然后,这些保守区之间、之前和之后的碱基的a、t、g或c随机变化,导致具有不同表达强度的启动子。或者,保守区发生改变而周围序列保持固定,这也导致具有不同强度的启动子。然后将这些序列混合物克隆(通过gibson组装、goldengate组装、cliva组装、lcr或限制性连接)(25,36,50,79)到翻译偶联的乳糖通透酶之前,得到可通过实施例5中所述的筛选方案筛选的乳糖通透酶的表达盒文库。
基于核心启动子创建真核启动子文库(13)。对于酿酒酵母,该启动子可以是异源tef启动子ptef1,其用uas序列增强并且被突变以改变启动子强度(12)。订购这样的启动子并如上所述将其克隆到翻译偶联的乳糖通透酶之前,并将最终构建体转化到酵母如酿酒酵母中,在质粒上或整合到染色体中。
非翻译区对于原核生物有核糖体结合位点组成,对于真核生物由kozak序列组成。使两个序列随机化并如上所述将其克隆到编码序列之前,得到具有不同翻译效率的表达盒文库。可以使用诸如计算序列翻译效率相关性的rbs计算器等工具使随机化合理化,并减少必须包括在文库中的变体的数量(67)。
通过改变基因的编码序列而不改变蛋白质的氨基酸序列来改变密码子使用。这被称为密码子适应。整个密码子序列中的密码子使用以这样的方式改变:在某些区域中引入更多或更少稀有密码子,导致表达效率和折叠效率改变。在其序列中仅具有稀有密码子(通过密码子使用数据库(61)在生物体基础上确定)的通透酶较具有完全密码子优化序列的通透酶(仅具有在靶生物体中具有高发生率的密码子)显示出更低的翻译速率。此外,编码序列的第一密码子也影响kozak或核糖体结合位点效率(62,69,78)。
通过数据库中存在的内源或外源转录终止子序列改变转录终止子区域(18,26)。这些转录终止子也类似于上述方法进行克隆。
实施例7:表达乳糖通透酶并且不导致乳糖杀伤的表达盒的富集
根据实施例3和7创建表达盒。这得到乳糖通透酶的表达盒的文库。根据实施例3和4的方法选择导致乳糖通透酶表达的表达盒。根据实施例2和5所述的方法选择不导致乳糖杀伤的表达乳糖通透酶的表达盒。通过测序和通过实施例2中所述的方法进一步分析所选择的表达盒。
实施例8:用来源于幽门螺杆菌(helicobacterpylori)的岩藻糖基转移酶与大肠杆菌发酵生产2-岩藻糖基乳糖
将突变株(其中敲除基因lacz、glgc、agp、pfka、pfkb、pgi、arca、iclr、wcaj并且如实施例1和实施例2中所述通过组成型表达表达lacy以确保在所有培养条件下的表达)用来源于幽门螺杆菌的岩藻糖基转移酶和来自青春双岐杆菌(bifidobacteriumadolescentis)的蔗糖磷酸化酶进一步转化,所述岩藻糖基转移酶和所述蔗糖磷酸化酶也是组成型表达的。组成型启动子来源于由demey等2007年所述的启动子文库。将该菌株在如材料和方法中所述但具有30g/l蔗糖和50g/l乳糖的培养基中进行培养。这导致形成高达1.5g/l的2’-岩藻糖基乳糖。
实施例9:用大肠杆菌补料分批生产2-岩藻糖基乳糖
通过上述基因工程方法构建具有以下基因型的突变株:δlaczyaδglgcδagpδpgiδpfka-p22-baspδpfkbδarcaδiclr::slδwcajδlonδadhe-p14-frk+pcxp14-ft_幽门螺杆菌(具有序列seqidn°6的载体,参见图10),其中如实施例1和实施例2中所述改变乳糖通透酶表达。启动子p22和p14来源于由demey等(29)构建的启动子文库,并且与aerts等(1)所述的方法类似地进行克隆。“::sl”标记无痕基因缺失,因此没有保留在染色体中的frt位点。
将该菌株在如以上材料和方法中所述的生物反应器中在具有30g/l蔗糖和50g/l乳糖的无机培养基中进行培养。在分批阶段后,向生物反应器供应500g/l蔗糖、50g/l乳糖和1g/l硫酸镁七水合物。这导致在上清液中积累27.5g/l的岩藻糖基乳糖。
实施例10:用大肠杆菌生产乳糖基n三糖
通过基因工程用上述方法构建突变株,所述突变株在pbr322质粒上表达udp-n-乙酰葡糖胺转移酶、蔗糖磷酸化酶和l-谷氨酰胺:d-果糖-6-磷酸转氨酶和葡糖胺尿苷酰转移酶,所述质粒各自具有来自demey等(29)的启动子文库的组成型启动子以及具有β-内酰胺酶选择标志物。将该载体在具有如上所述组成性表达乳糖通透酶的基因型δlaczyaδglgcδagp::p14-frk-p22-baspδpgiδpfkaδpfkbδnagabcdeδmanaδnanatekδmanxyz的大肠杆菌突变株中转化。将该菌株在如上所述具有乳糖和蔗糖作为碳源的有或没有另外的甘油的摇瓶中进行培养。该生产宿主不经历乳糖杀伤并且由添加的乳糖分别产生62.5mg/l和55.3mg/l乳糖基n三糖。
实施例11:在rdna基因座处构建马克斯克鲁维酵母乳糖通透酶(p1)敲入
首先,质粒p2a_2μ_lac4用于转化到酿酒酵母by4742野生型菌株中。使用gietz方案(40)使用总计4μg的质粒进行转化。将转化的酵母细胞铺在sd-csm漏失板(不含尿嘧啶)上。两天后,在板上观察到生长,并对酵母菌落中期望质粒的存在进行测试。在所有33个菌落上进行菌落pcr。所有菌落对质粒p2a_2μ_lac4的存在测试为阳性。选择菌落5用于进一步使用。用2μg获得自pjet_ki_p1_lac12_t@rdna的双链线性dna转化该菌落。将转化的细胞铺在具有乳糖作为唯一c源的sd-csm-ura板上。转化后3天,使几个菌落充分生长以测试酵母属rdna中lac12表达盒的存在。对所有菌落进行菌落pcr。所有菌落对lac12表达盒的存在测试为阳性。选择菌落6用于进一步使用(酵母菌ki_p1_lac12_t@rdna)。
实施例12:在rdna基因座处构建马克斯克鲁维酵母乳糖通透酶(p2)敲入
首先,质粒p2a_2μ_lac4用于转化到酿酒酵母by4742野生型菌株中。使用gietz方案(40)使用总计4μg的质粒进行转化。将转化的酵母细胞铺在sd-csm漏失板(不含尿嘧啶)上。两天后,在板上观察到生长,并对酵母菌落中期望质粒的存在进行测试。在所有33个菌落上进行菌落pcr。所有菌落对质粒p2a_2μ_lac4的存在测试为阳性。选择菌落5用于进一步使用。用2μg获得自pjet_ki_p2_lac12_t@rdna的双链线性dna转化该菌落。将转化的细胞铺在具有乳糖作为唯一c源的sd-csm-ura板上。转化后3天,使几个菌落充分生长以测试酵母属rdna中lac12表达盒的存在。对所有菌落进行菌落pcr。所有菌落对lac12表达盒的存在测试为阳性。选择菌落7用于进一步使用(酵母属ki_p2_lac12_t@rdna)。
实施例13:野生型酵母菌株和2种突变酵母菌株在乳糖上的生长
用野生型克鲁维酵母、酵母菌ki_p1_lac12_t@rdna和酵母菌ki_p2_lac12_t@rdna建立如材料和方法中所述的摇瓶实验。这些菌株在包含乳糖(20g/l)作为唯一c源的摇瓶培养基中生长。从表1中可以看出,突变酿酒酵母菌株与野生型乳酸马克斯克鲁维酵母生长一样快,所述乳酸马克斯克鲁维酵母已知在乳糖上快速生长(31)。因此,组成型表达的乳糖通透酶确保了酵母细胞中乳糖的快速且有效流入。
表1
实施例14:乳糖对野生型酵母菌株和不经历乳糖杀伤的突变酵母菌株的影响
用野生型克鲁维酵母、酵母菌ki_p1_lac12_t@rdna和酵母菌ki_p2_lac12_t@rdna建立如材料和方法中所述的摇瓶实验。这些菌株在葡萄糖摇瓶培养基(20g/l葡萄糖,如材料和方法中所述)中生长,并且在指数期中期添加乳糖(添加200g/l储液,导致10g/l的最终浓度)。从图11和图12中可以看出,突变株不经历乳糖杀伤。然而,在实施例13中证明了这些酵母细胞中乳糖的快速且有效流入。
实施例15:不经历乳糖杀伤的乳糖通透酶表达盒的测序
对来源于上述筛选的46个菌落进行测序,得到seqidn°12-57(图18)。这些序列是当在大肠杆菌中表达时不导致乳糖杀伤的启动子和rbs变体。注意,这是对大量已被测序的菌落的选择,因此,筛选方法已导致较图18中所示的多得多的序列,并且如上所述的替代文库创建方法也将导致除图18中所示的那些之外的其他序列。
实施例16:乳糖通透酶表达盒突变株的μmax的测定
所有菌株从lb-琼脂板开始或从冷冻管开始并接种在5.0mlluria肉汤培养基(10g胰蛋白胨;5g酵母提取物;10gnacl)中。在37℃下过夜(o/n)生长后,将1ml该预培养物添加到包含100mlph7.0基本乳糖培养基(2.0g/lnh4cl;5.0g/l;(nh4)2so4;3.0g/lkh2po4.7.3g/lk2hpo4;8.4g/lmops;0.5g/lmgso4×7h2o;0.5g/lnacl;10g/l乳糖;0.4mg/lna2edta×2h2o;0.03mg/lh3bo3;1.01mg/l硫胺素hcl;0.94mg/lzncl2;0.5mg/lcocl2×6h2o;0.38mg/lcucl2×2h2o;1.59mg/lmncl2×4h2o;3.6mg/lcacl2和0.096mg/lna2moo4×2h2o)的500ml摇瓶中。在37℃下过夜生长后,将两种预培养物用基本乳糖培养基稀释至od600为0.050。然后将悬液转移至96mtp板(n=32),用易密封盖覆盖。使用infinitem200pro(tecan)在以下条件下每10分钟进行od测量持续24小时:温度:37℃+/-0.5;摇动597秒;2mm摇动幅度;280rpm;波长od600;闪光#10;停止时间150毫秒)。
实施例17:乳糖通透酶表达盒的表征
减少或消除乳糖杀伤的一种方法是显著降低或消除乳糖通透酶的活性(参见实施例16)。然而,鉴于生产基于乳糖或半乳糖的生物产品,需要进入细胞中的高乳糖通量。在此,证明了乳糖杀伤抗性突变株的乳糖通量与经历乳糖杀伤的野生型生物体的乳糖通量相比仍然可比、相等或者甚至更高。为此,将新的乳糖通透酶表达盒引入到仍然表达β-半乳糖苷酶的mg1655δlacy菌株中。这些新菌株的生长速率是乳糖通量的度量,因为在乳糖通透酶表达中具有显著降低的表达的任何菌株将具有显著降低的生长速率。
该分析的结果示于图19中。该图中示出的几乎所有菌株具有等于或高于野生型的生长速率,表明等于或高于野生型的乳糖通透酶活性和表达,然而,与野生型表达系统形成对比,这些表达盒不导致乳糖杀伤。然而,该方法受β-半乳糖苷酶基因表达(其成为生长的限速步骤)限制。因此,通过上述的上述翻译偶联系统测量乳糖通透酶基因的表达。氯霉素的最小抑制浓度(minimalinhibitionconcentration,mic)指示乳糖通透酶基因的表达,并且对于每个盒测定最小抑制浓度。使用与野生型相比仍然导致相同生长速率的最低mic作为表达野生型乳糖通透酶表达的指示。
与野生型菌株相同的生长速率的具有最低mic的盒具有约20mg/l氯霉素的mic。具有略低生长速率的突变株具有15mg/l至20mg/l的mic,并且具有相等或更高生长速率的突变株具有20mg/l至80mg/l的mic。85%的序列属于后一种,这意味着鉴定为乳糖杀伤阴性表达盒的大多数序列具有较高的乳糖通透酶表达,与文献中先前描述的相反。
实施例18:laciq启动子表达盒的构建
与上述方法类似,将placiq启动子克隆到大肠杆菌lacy基因之前。该构建体的最终序列示于图20中。
实施例19:经历乳糖杀伤的本领域中使用的其他启动子
启动子placiq是本领域中用于在大肠杆菌中表达乳糖通透酶的启动子。该启动子的使用导致乳糖摄取到细胞中。然而,未测试这样的摄取的效果。野生型在乳糖上的生长速率与laciq启动子(μmax分别为0.1和0.11小时-1)没有显著差异,因此表明类似的乳糖摄取速率。该laciq表达盒的乳糖杀伤筛选证明该启动子也导致乳糖杀伤(参见图21)。这证明存在导致乳糖杀伤的特定启动子序列和不导致乳糖杀伤的其他序列。序列的合理化是不可能的,并且单个序列的反复试验是一项费力的工作,这将导致巨大的成本,因此,上述用于鉴定不经历乳糖杀伤的表达盒的方法是避免冗长且昂贵的筛选工作的完美方法。
实施例20:用酿酒酵母生产乳糖基n四糖
例如如由lee等(52)所述,将实施例11和12中构建的突变株进一步用来源于脑膜炎奈瑟菌(neisseriameningitidis)的β-1,4-半乳糖基转移酶和β-1,3-n-乙酰葡糖胺转移酶使用标准酵母表达载体转化,所述酶也是组成型表达的。将菌株培养在如材料和方法中所述但具有20g/l蔗糖和20g/l乳糖的培养基中。这导致形成高至30mg/l的乳糖基n四糖。
实施例21:用酿酒酵母生产2-岩藻糖基乳糖
例如如由lee等(52)所述,将实施例11和12中构建的突变株进一步用大肠杆菌的gdp-岩藻糖合酶和gdp-甘露糖4,6-脱水酶和来源于幽门螺杆菌的岩藻糖基转移酶使用标准酵母表达载体转化,所述酶也是组成型表达的。将菌株培养在如材料和方法中所述但具有20g/l蔗糖和20g/l乳糖的培养基中。这导致形成高至10mg/l的2岩藻糖基乳糖。
参考文献:
1.aerts,d.,t.verhaeghe,m.demey,t.desmet,andw.soetaert.2010.aconstitutiveexpressionsystemforhighthroughputscreening.engineeringinlifesciences10:doi:10.1002/elsc.201000065.
2.agrawal,n.,p.v.n.dasaradhi,a.mohmmed,p.malhotra,r.k.bhatnagar,ands.k.mukherjee.2003.rnainterference:biology,mechanism,andapplications.microbiologyandmolecularbiologyreviews67:657-685.
3.alper,h.,c.fischer,e.nevoigt,andg.stephanopoulos.2005.tuninggeneticcontrolthroughpromoterengineering.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica102:12678-12683.
4.avihoo,a.,i.gabdank,m.shapira,andd.barash.2007.insilicodesignofsmallrnaswitches.ieeetransactionsonnanobioscience6:4-11.
5.ayres,e.k.,v.j.thomson,g.merino,d.balderes,andd.h.figurski.1993.precisedeletionsinlargebacterialgenomesbyvector-mediatedexcision(vex):thetrfageneofpromiscuousplasmidrk2isessentialforreplicationinseveralgram-negativehosts.journalofmolecularbiology230:174-185.
6.balbás,p.,m.alexeyev,i.shokolenko,f.bolivar,andf.valle.1996.apbrintfamilyofplasmidsforintegrationofcloneddnaintotheescherichiacolichromosome.gene172:65-69.
7.balbas,p.,andg.gosset.2001.chromosomaleditinginescherichiacoli.molecularbiotechnology19:1-12.
8.barrett,a.r.,y.kang,k.s.inamasu,m.s.son,j.m.vukovich,andt.t.hoang.2008.genetictoolsforallelicreplacementinburkholderiaspecies.appliedandenvironmentalmicrobiology74:4498-4508.
9.beauprez,j.2010.metabolicmodellingandengineeringofescherichiacoliforsuccinateproduction.phd.ghentuniversity,ghent.
10.beekwilder,j.,h.m.vanrossum,f.koopman,f.sonntag,m.buchhaupt,j.schrader,r.d.hall,d.bosch,j.t.pronk,a.j.a.vanmaris,andj.-m.daran.2014.polycistronicexpressionofaβ-carotenebiosyntheticpathwayinsaccharomycescerevisiaecoupledtoβ-iononeproduction.
11.biofuge,h.thermo.
12.blazeck,j.,r.garg,b.reed,andh.s.alper.2012.controllingpromoterstrengthandregulationinsaccharomycescerevisiaeusingsynthetichybridpromoters.biotechnologyandbioengineering109:2884-2895.
13.blount,b.a.,t.weenink,s.vasylechko,andt.ellis.2012.rationaldiversificationofapromoterprovidingfine-tunedexpressionandorthogonalregulationforsyntheticbiology.plosone7:e33279.
14.bode,l.2012.humanmilkoligosaccharides:everybabyneedsasugarmama.glycobiology22:1147-1162.
15.bode,l.2006.recentadvancesonstructure,metabolism,andfunctionofhumanmilkoligosaccharides.thejournalofnutrition136:2127-2130.
16.brendadatabase2006,postingdate.[online.]
17.cabantous,s.,t.c.terwilliger,andg.s.waldo.2005.proteintagginganddetectionwithengineeredself-assemblingfragmentsofgreenfluorescentprotein.natbiotech23:102-107.
18.cambray,g.,j.c.guimaraes,v.k.mutalik,c.lam,q.-a.mai,t.thimmaiah,j.m.carothers,a.p.arkin,andd.endy.2013.measurementandmodelingofintrinsictranscriptionterminators.nucleicacidsresearch41:5139-5148.
19.canton,b.,a.labno,andd.endy.2008.refinementandstandardizationofsyntheticbiologicalpartsanddevices.natbiotech26:787-793.
20.caspi,r.,t.altman,r.billington,k.dreher,h.foerster,c.a.fulcher,t.a.holland,l.m.keseler,a.kothari,a.kubo,m.krummenacker,m.latendresse,l.a.mueller,q.ong,s.paley,p.subhraveti,d.s.weaver,d.weerasinghe,p.zhang,andp.d.karp.2014.themetacycdatabaseofmetabolicpathwaysandenzymesandthebiocyccollectionofpathway/genomedatabases.nucleicacidsresearch42:d459-d471.
21.chen,x.,anda.varki.2009.advancesinthebiologyandchemistryofsialicacids.acschemicalbiology5:163-176.
22.cherepanov,p.p.,andw.wackernagel.1995.genedisruptioninescherichiacoli:tcrandkmrcassetteswiththeoptionofflp-catalyzedexcisionoftheantibiotic-resistancedeterminant.gene158:9-14.
23.consortium,t.u.2015.uniprot:ahubforproteininformation.nucleicacidsresearch43:d204-d212.
24.coppa,g.v.,l.zampini,t.galeazzi,ando.gabrielli.2006.prebioticsinhumanmilk:areview.digestiveandliverdisease38:s291-s294.
25.coussement,p.,j.maertens,j.beauprez,w.vanbellegem,andm.demey.2014.onestepdnaassemblyforcombinatorialmetabolicengineering,p.70-77,vol.23.
26.curran,k.a.,a.s.karim,a.gupta,andh.s.alper.2013.useofexpression-enhancingterminatorsinsaccharomycescerevisiaetoincreasemrnahalf-lifeandimprovegeneexpressioncontrolformetabolicengineeringapplications,p.88-97,vol.19.
27.datsenko,k.a.,andb.l.wanner.2000.one-stepinactivationofchromosomalgenesinescherichiacolik-12usingpcrproducts.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica97:6640-6645.
28.davis,s.s.1997.biomedicalapplicationsofnanotechnology-implicationsfordrugtargetingandgenetherapy.trendsinbiotechnology15:217-224.
29.demey,m.,j.maertens,g.j.lequeux,w.k.soetaert,ande.j.vandamme.2007.constructionandmodel-basedanalysisofapromoterlibraryfore.coii:anindispensabletoolformetabolicengineering.bmcbiotechnology7:34-48.
30.dicarlo,j.e.,j.e.norville,p.mali,x.rios,j.aach,andg.m.church.2013.genomeengineeringinsaccharomycescerevisiaeusingcrispr-cassystems.nucleicacidsresearch.
31.drozdiková,e.,m.garaiová,z.csáky,m.obernauerová,andl.hapala.2015.productionofsqualenebylactose-fermentingyeastkluyveromyceslactiswithreducedsqualeneepoxidaseactivity.lettersinappliedmicrobiology61:77-84.
32.dumon,c.,b.priem,s.l.martin,a.heyraud,c.bosso,ande.samain.2001.invivofucosylationoflacton-neotetraoseandlacton-neohexaosebyheterologousexpressionofhelicobacterpyloriα-1,3fucosyltransferaseinengineeredescherichiacoli.glycoconjugatejournal18:465-474.
33.dykhuizen,d.,andd.hartl.1978.transportbythelactosepermeaseofescherichiacoliasthebasisoflactosekilling.journalofbacteriology135:876-882.
34.eames,m.,andt.kortemme.2012.cost-benefittradeoffsinengineeredlacoperons.science336:911-915.
35.edwards,j.s.,r.ramakrishna,c.h.schilling,andb.o.palsson.1999.metabolicfluxbalanceanalysis.metabolicengineering:13-57.
36.engler,c.,r.gruetzner,r.kandzia,ands.marillonnet.2009.goldengateshuffling:aone-potdnashufflingmethodbasedontypellsrestrictionenzymes.plosone4:e5553.
37.filonov,g.s.,j.d.moon,n.svensen,ands.r.jaffrey.2014.broccoli:rapidselectionofanrnamimicofgreenfluorescentproteinbyfluorescence-basedselectionanddirectedevolution.journaloftheamericanchemicalsociety.
38.galperin,m.y.,andg.r.cochrane.2009.nucleicacidsresearchannualdatabaseissueandthenaronlinemolecularbiologydatabasecollectionin2009.nucleicacidresearch37:d1-4.
39.ghazi,a.,h.therisod,ande.shechter.1983.comparisonoflactoseuptakeinrestingandenergizedescherichiacolicells:highratesofrespirationinactivatethelaccarrier.journalofbacteriology154:92-103.
40.gietz,r.d.,r.h.schiestl,a.r.willems,andr.a.woods.1995.studiesonthetransformationofintactyeastcellsbytheliac/ss-dna/pegprocedure.yeast11:355-360.
41.hammer,k.,i.mijakovic,andp.r.jensen.2006.syntheticpromoterlibraries-tuningofgeneexpression.trendsinbiotechnology24:53-55.
42.hanahan,d.,j.jessee,andf.r.bloom.1991.plasmidtransformationofescherichiacoiiandotherbacteria.methodsinenzymology204:63-113.
43.hebert,c.g.,j.j.valdes,andw.e.bentley.2008.beyondsilencing--engineeringapplicationsofrnainterferenceandantisensetechnologyforalteringcellularphenotype.currentopinioninbiotechnology19:500-505.
44.heinemann,m.,a.kummel,r.ruinatscha,ands.panke.2005.kegg:kyotoencyclopediaofgenesandgenomesinsilicogenome-scalereconstructionandvalidationofthestaphylococcusaureusmetabolicnetwork.biotechnolbioeng92:850-864.
45.hoang,t.t.,r.r.karkhoff-schweizer,a.j.kutchma,andh.p.schweizer.1998.abroad-host-rangefip-frtrecombinationsystemforsite-specificexcisionofchromosomally-locateddnasequences:applicationforisolationofunmarkedpseudomonasaeruginosamutants.gene212:77-86.
46.jiang,w.,d.bikard,d.cox,f.zhang,andl.a.marraffini.2013.rna-guidededitingofbacterialgenomesusingcrispr-cassystems.natbiotech31:233-239.
47.kanehisa,m.,m.araki,s.goto,m.hattori,m.hirakawa,m.itoh,t.katayama,s.kawashima,s.okuda,t.tokimatsu,andy.yamanishi.2008.keggforlinkinggenomestolifeandtheenvironment.nucleicacidresearch36:d480-484.
48.koizumi,s.,t.endo,k.tabata,anda.ozaki.1998.large-scaleproductionofudp-galactoseandglobotriosebycouplingmetabolicallyengineeredbacteria.natbiotech116:847-850.
49.kojima,k.k.,t.matsumoto,andh.fujiwara.2005.eukaryotictranslationalcouplinginuaaugstop-startcodonsforthebicistronicrnatranslationofthenon-longterminalrepeatretrotransposonsart1.molecularandcellularbiology25:7675-7686.
50.kok,s.d.,l.h.stanton,t.slaby,m.durot,v.f.holmes,k.g.patel,d.platt,e.b.shapland,z.serber,j.dean,j.d.newman,ands.s.chandran.2014.rapidandreliablednaassemblyvialigasecyclingreaction.acssyntheticbiology3:97-106.
51.kristensen,c.s.,l.eberl,j.m.sanchez-romero,m.givskov,s.molin,andv.delorenzo.1995.site-specificdeletionsofchromosomallylocateddnasegmentswiththemultimerresolutionsystemofbroad-host-rangeplasmidrp4.journalofbacteriology1177:52-58.
52.lee,m.e.,w.c.deloache,b.cervantes,andj.e.dueber.2015.ahighlycharacterizedyeasttoolkitformodular,multipartassembly.acssyntheticbiology4:975-986.
53.levin-karp,a.,u.barenholz,t.bareia,m.dayagi,l.zelcbuch,n.antonovsky,e.noor,andr.milo.2013.quantifyingtranslationalcouplingine.colisyntheticoperonsusingrbsmodulationandfluorescentreporters.acssyntheticbiology2:327-336.
54.llaneras,f.,andj.picó.2008.stoichiometricmodellingofcellmetabolism.journalofbioscienceandbioengineering105:1-11.
55.lodi,t.,andc.donnini.2005.lactose-inducedcelldeathofβ-galactosidasemutantsinkluyveromyceslactis.femsyeastresearch5:727-734.
56.martin,p.,o.albagli,m.poggi,k.boulukos,andp.pognonec.2006.developmentofanewbicistronicretroviralvectorwithstrongiresactivity.bmcbiotechnology6:4.
57.mcshan,d.c.,s.rao,andi.shah.2003.pathminer:predictingmetabolicpathwaysbyheuristicsearch.bioinformatics19:1692-1698.
58.mendez-perez,d.,s.gunasekaran,v.j.orler,andb.f.pfleger.2012.atranslation-couplingdnacassetteformonitoringproteintranslationinescherichiacoli.metabolicengineering14:298-305.
59.merighi,m.,j.m.mccoy,andm.i.heidtman.2012.biosynthesisofhumanmilkoligosaccharidesinengineeredbacteria.wo2012112777
60.mutalik,v.k.,j.c.guimaraes,g.cambray,c.lam,m.j.christoffersen,q.-a.mai,a.b.tran,m.paull,j.d.keasling,a.p.arkin,andd.endy.2013.preciseandreliablegeneexpressionviastandardtranscriptionandtranslationinitiationelements.natmeth10:354-360.
61.nakamura,y.,t.gojobori,andt.ikemura.2000.codonusagetabulatedfrominternationaldnasequencedatabases:statusfortheyear2000.nucleicacidsresearch28:292.
62.pechmann,s.,andj.frydman.2012.evolutionaryconservationofcodonoptimalityrevealshiddensignaturesofcotranslationalfolding.natstructmolbiol20:237-243.
63.peijnenburg,a.c.m.,g.venema,ands.bron.1990.translationalcouplinginapenp-laczgenefusioninbacillussubtilisandescherichiacoli:useofauaasarestartcodon.1990221:267-272.
64.pothoulakis,g.,f.ceroni,b.reeve,andt.ellis.2014.thespinachrnaaptamerasacharacterizationtoolforsyntheticbiology.acssyntheticbiology3:182-187.
65.rasmussen,l.,h.sperling-petersen,andk.mortensen.2007.hittingbacteriaattheheartofthecentraldogma:sequence-specificinhibition.microbialcellfactories6:24.
66.rhodius,v.a.,v.k.mutalik,andc.a.gross.predictingthestrengthofup-elementsandfull-lengthe.colisigmaepromoters.nucleicacidsresearch40:2907-2924.
67.salis,h.m.,e.a.mirsky,andc.a.voigt.2009.automateddesignofsyntheticribosomebindingsitestocontrolproteinexpression.natbiotech27:946-950.
68.seed,b.,andj.holgersson.1999.fucosyltransferasegenesandusesthereofpatentus5858752
69.supek,f.,\#352,andt.muc.2010.onrelevanceofcodonusagetoexpressionofsyntheticandnaturalgenesinescherichiacoli.genetics185:1129-1134.
70.timblin,c.r.,andm.l.kahn.1984.lactoseinhibitsthegrowthofrhizobiummeliloticellsthatcontainanactivelyexpressedescherichiacolilactoseoperon.journalofbacteriology158:1204-1207.
71.varki,a.1992.diversityinthesialicacids.glycobiology2:25-40.
72.waldo,g.s.,b.m.standish,j.berendzen,andt.c.terwilliger.1999.rapidprotein-foldingassayusinggreenfluorescentprotein.natbiotech17:691-695.
73.welch,m.,s.govindarajan,j.e.ness,a.villalobos,a.gurney,j.minshull,andc.gustafsson.2009.designparameterstocontrolsyntheticgeneexpressioninescherichiacoli.plosone4:e7002.
74.williams,j.,j.luke,andc.hodgson.2009.strainengineeringbygenomemasstransfer:efficientchromosomaltraittransfermethodutilizingdonorgenomicdnaandrecipientrecombineeringhosts.molecularbiotechnology43:41-51.
75.wilson,d.m.,r.m.putzrath,andt.h.wilson.1981.inhibitionofgrowthofescherichiacolibylactoseandothergalactosides.biochimicaetbiophysicaacta(bba)-biomembranes649:377-384.
76.xu,x.j.,l.m.cao,andx.chen.2008.elementaryfluxmodeanalysisforoptimizedethanolyieldinanaerobicfermentationofglucosewithsaccharomycescerevisiae.chinesejournalofchemicalengineering16:135-142.
77.yanase,h.,j.kurii,andk.tonomura.1988.fermentationoflactosebyzymomonasmobiliscarryingalac+recombinantplasmid.journaloffermentationtechnology66:409-415.
78.zhou,m.,j.guo,j.cha,m.chae,s.chen,j.m.barral,m.s.sachs,andy.liu.2013.non-optimalcodonusageaffectsexpression,structureandfunctionofclockproteinfrq.nature495:111-115.
79.zou,r.,k.zhou,g.stephanopoulos,andh.p.too.2013.combinatorialengineeringof1-deoxy-d-xylulose5-phosphatepathwayusingcross-lapping<italic>invitro</italic>assembly(cliva)method.plosone8:e79557.
- 还没有人留言评论。精彩留言会获得点赞!