甲氨基阿维菌素苯甲酸盐的制备方法与流程
本发明属于农业有机化学领域,特别涉及甲氨基阿维菌素苯甲酸盐的制备方法。
背景技术:
甲氨基阿维菌素苯甲酸盐是一种高效、低毒、低残留的绿色环保型生物源杀虫剂,其分子式为C49H75NO13·C7H6O2(Bla),C48H73NO13·C7H6O2(B1b)。原药为白色或淡黄色结晶粉末;熔点141~146℃,易溶于丙酮、甲醇、乙醇、异丙醇等有机溶剂;pH值为7.0时,在水中溶解度为2.4×10-2g·L-1,pH值为5.0时,在水中溶解度为0.3×10-2g·L-1。
甲维盐是生物发酵产物阿维菌素的衍生物,与阿维菌素相比,其生物活性提高了约3个数量级。对鳞翅目昆虫的幼虫和其它许多害虫的活性极高,在非常低的剂量(0.084-2g/ha)下即具有很好的效果,而且在防治害虫的过程中对益虫没有伤害,有利于对害虫的综合防治,另外扩大了杀虫谱,降低了对人畜的毒性。目前,甲维盐已成为国内农药市场中的一线产品。
目前国内甲维盐的合成主要采用两步法合成工艺(CN201310368725.1)。该工艺虽然已经成熟,但是仍然存在不足之处:
(1)以氯甲酸烯丙酯保护5-位羟基,在脱保护时需要用到催化剂四(三苯基磷)钯,该催化剂价格昂贵,大大地增加了生产成本;
(2)氧化过程中以二甲基亚砜作为氧化剂,产生的杂质导致四(三苯基磷)钯的活性降低,大大地增加了四(三苯基磷)钯的用量;
(3)生产过程中需要使用到醋酸异丙酯作为反应溶剂,使用量很大,醋酸异丙酯价格昂贵,使甲维盐生产成本偏高;
(4)生产过程中,由于中间需要更换溶剂,产物需要做处理,增加了生产时间、溶剂的消耗、后处理所产生的废液及人工费,增加了甲维盐生产成本。
虽然传统工艺在操作中比较简单,生产条件容易达到,但该生产路线产品收率较低、纯度不高、成本高,很难满足市场对产品的需求。
由于上述缺点的存在,不利于甲维盐大规模工业化生产。
技术实现要素:
本发明的目的是提供一种甲氨基阿维菌素苯甲酸盐的制备方法,具体技术方案如下:
甲氨基阿维菌素苯甲酸盐的制备方法,包括以下步骤:
(1)单保护:在有机碱和有机试剂的存在下,式F所示的阿维菌素和酰化试剂反应,然后将pH值调整为6-7,萃取,得到式B所示化合物;
式F
式B
(2)氧化:在有机试剂的存在下,式B所示化合物和复合氧化试剂反应,使C5-羟基氧化成羰基,反应完成后,将pH值调整为6-7,萃取,得到式C所示化合物;
式C
(3)氨化:在有机溶剂的存在下,将式C所示化合物在加入催化剂Ⅱ的条件下与氨化试剂Ⅰ反应,将式C所示化合物中C4上的羰基氨化为亚甲氨基,再加入试剂Ⅲ和氨化还原剂Ⅳ,使得C4上亚甲氨基还原成甲氨基,反应完成后,将pH值调整为6-7,萃取,得到式D所示化合物;
式D
(4)脱保护:在有机溶剂中将式D所示化合物与锌粉、氯化铵反应,使得式D所示化合物中C5上的2-硝基苯乙酰基脱除为羟基,得到式E所示化合物;
式E
(5)成盐:将步骤(4)脱保护后得到的物料进行过滤、水洗、分离,得到式E物质的有机相溶液,向该有机相溶液中加入苯甲酸,搅拌反应成盐后,去除有机溶剂,即得甲氨基阿维菌素苯甲酸盐。
其中,相对于1摩尔的式F所示的物质,步骤(1)中,2-硝基苯乙酰氯的用量可以为1-5摩尔,优选为1.05-3摩尔,所述有机碱的用量为1-10摩尔,优选为1.05-5摩尔,更优选为1.2-3摩尔;步骤(2)中,所述式F与复合氧化试剂的质量比15~30:13,优选质量比为20~25:13;步骤(3)中,所述氨化试剂Ⅰ的用量可以为1.5-50摩尔,优选为3-20摩尔,更优选为5-10摩尔,所述催化剂Ⅱ的用量可以为0.0001-0.5重量份,优选为0.001-0.3重量份,更优选为0.01-0.2重量份,特别优选为0.05-0.15重量份;所述试剂Ⅲ的用量为1-5重量份,优选为1.5-2.5重量份;所述氨化还原剂Ⅳ的用量,为0.0001-0.5重量份,优选为0.001-0.3重量份,更优选为0.01-0.1重量;步骤(4)所述锌粉的用量为1~5摩尔,优选为1.1~2.5摩尔;所述氯化铵的用量为0.8~3摩尔,优选为1~1.5摩尔;步骤(5)中,苯甲酸的用量可以为1-10摩尔,优选为1.02-5摩尔,更优选为1.05-3摩尔。
其中,步骤(1)中,所述酰化反应条件包括:温度可以为-50℃至-2℃,优选为-25℃至-15℃,时间可以为0.5-5小时,优选为0.8-2小时;步骤(2)中,所述氧化化反应条件包括:温度可以为-30℃至0℃,优选为-20℃至-10℃,时间可以为0.5-2小时,优选为0.6-1小时;步骤(3)中,所述氨化、还原反应条件包括:温度可以为0℃-40℃,优选为5℃-25℃,时间可以为1-20小时,优选为4-6小时;步骤(4)中,所述脱保护反应条件包括:温度可以为10℃-30℃,优选为15℃-25℃,时间可以为2-20小时,优选为4-10小时;步骤(5)中,所述成盐反应条件包括:温度可以为0℃-50℃,时间可以为0.1-5小时。
其中,步骤(2)中,所述的复合氧化试剂是由二甲基亚砜、四甲基乙二胺和磷酸苯酯二酰氯组成的,且二甲基亚砜与四甲基乙二胺的质量比为1.4:1.0;二甲基亚砜与磷酸苯酯二酰氯的质量比为1.3:1.0。步骤(3)中,所述的氨化试剂Ⅰ为七甲基二硅氮烷或甲胺-甲醇溶液;所述的催化剂Ⅱ为氯化锌、对甲苯磺酸锌或三氟乙酸锌;所述的试剂Ⅲ为乙醇或甲醇;所述的氨化还原剂Ⅳ为硼氢化钠。
其中,步骤(1)、步骤(2)、步骤(3)、步骤(4)中,所述有机溶剂选自乙醇、二氯甲烷、正己烷、苯、甲苯、三氯甲烷、四氯甲烷、石油醚、氯苯、二氧六环和四氢呋喃中的至少一种,优选二氯甲烷、三氯甲烷和四氢呋喃中的至少一种,最优选为二氯甲烷;所述有机溶剂的用量可以为1-100L,优选为2-10L,更优选为3-5L。
其中,步骤(1)、步骤(2)、步骤(3)所述调整pH值为6-7所用的酸可以是常用的各种浓度的无机酸,例如磷酸、盐酸、硫酸和硝酸中的至少一种,优选浓度为5%-20%重量的硫酸、浓度为5%-20%重量的盐酸和浓度为5%-20%重量的磷酸中的至少一种,更优选浓度为8%-12%重量的盐酸。
其中,步骤(1)、步骤(2)、步骤(3)、步骤(5)中,水洗所用水量和水洗次数没有特别的要求,水洗所用水量可以为还原反应后物料体积的1-5倍;水洗次数可以为1-5次。水洗后的物料可以进行常规的干燥,例如加入无水硫酸钠进行干燥。
其中,步骤(5)的目的是将式E的物质与苯甲酸形成苯甲酸盐。其中,在成盐反应条件下接触后的物料中含有甲维盐,可以通过常规的纯化方式如蒸馏,除去有机溶剂,得到甲维盐。
此外,式F所示的阿维菌素包括阿维菌素B1a和阿维菌素B1b,阿维菌素B1a中R代表乙基,阿维菌素B1b中R代表甲基。式F所示的阿维菌素中,阿维菌素B1a和阿维菌素B1b的重量比可以为任意比例,优选为1:(10-40),更优选为1:(15-25)。
本发明有以下优点:(1)在单保护中使用2-硝基苯乙酰氯作为保护基,脱保护中避免了使用催化剂四(三苯基磷)钯,也避免了氧化试剂二甲基亚砜对催化剂四(三苯基磷)钯所产生的影响,从而减少了生产成本;(2)生产中在以阿维菌素为起始原料,经过单保护-氧化-氨化-脱保护-苯甲酸成盐五步反应的过程中,都使用相同的溶剂,不再更换溶剂,从而提高了产品生产效率,降低了生产成本;(3)生产可以使用一锅法,即单保护-氧化-氨化-脱保护四步反应的过程中均在同一体系里完成,中间不需对产物进行后处理,有效地缩短了生产时间,减少了溶剂的消耗、后处理所产生的废液及人工费;(4)本发明简化了生产工艺,操作条件简单,提高了产品的收率及纯度,降低了生产成本。
附图说明
图1是实施例1步骤(1)的萃余有机相物料中式B物质的质谱图。
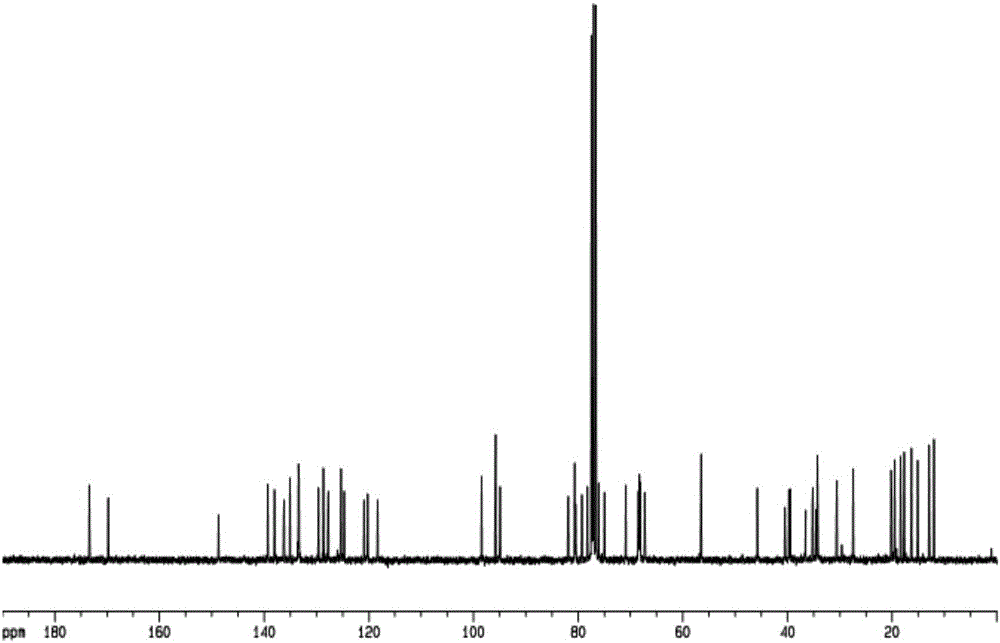
图2是实施例1步骤(1)的萃余有机相物料中式B物质的碳谱图。
图3是实施例1步骤(2)的萃余有机相物料中式C物质的质谱图。
图4是实施例1步骤(2)的萃余有机相物料中式C物质的碳谱图。
图5是实施例1步骤(3)的萃余有机相物料中式D物质的质谱图。
图6是实施例1步骤(3)的萃余有机相物料中式D物质的氢谱图。
图7是实施例1步骤(4)的萃余有机相物料中式E物质的质谱图。
图8是实施例1步骤(5)得到的产品中的甲维盐的氢谱图。
图9是实施例1步骤(5)得到的产品中的甲维盐的碳谱图。
图10是实施例1步骤(5)得到的产品中的甲维盐的HPLC图。
具体实施方式
本发明中,在未作相反说明的情况下,使用的气体和液体的体积数值为25℃、一个标准大气压下的数值;所用的试剂和装置均为市售品,所用试剂的纯度为分析纯。
实施例1
(1)向1000mL三口瓶中加入100g(0.115摩尔)的阿维菌素(购自湖北远成赛创科技有限公司,如式F所示的物质,阿维菌素B1a和阿维菌素B1b的重量比为1:20)和400mL无水二氯甲烷,溶解充分后,降温到-15℃,加入有机碱四甲基乙二胺24g(0.206摩尔),降温到-20℃,缓慢滴加酰化试剂2-硝基苯乙酰氯26g(0.217摩尔)与无水二氯甲烷50ml的混合溶液,在-20℃下反应1小时,使得式F中的C4-羟基中的氢替换为2-硝基苯乙酰基,反应完成后,将反应后物料的pH值调整为6-7,用水萃取,除水后减压蒸馏,得到式B阿维菌素C4-2-硝基苯乙酰基,C5-羟基的单保护产物物料。
(2)将步骤(1)得到的式B所示物质溶于350mL无水二氯甲烷,溶解充分后,降温到-20℃,再加入复合氧化试剂65g(二甲基亚砜、四甲基乙二胺和磷酸苯酯二酰氯组成)。在-20℃下反应1小时,使得式B中的C5-羟基氧化成羰基;反应完成后,将得到反应物料的pH值调整为6-7,用水萃取,除去水后减压蒸馏,得到式C阿维菌素C4-2-硝基苯乙酰基,C5-羰基的氧化产物。
(3)将步骤(2)得到的式C所示物质溶于250mL无水二氯甲烷,溶解充分后,降温到10℃,加入氨化试剂Ⅰ七甲基二硅氮烷102g(0.581摩尔)和催化剂Ⅱ氯化锌12g(0.12重量份),在10℃下反应2小时,使得式C中的C4上的羰基氨化为亚甲氨基。加入试剂Ⅲ甲醇150g(1.5重量份)和氨化还原剂Ⅳ硼氢化钠8g(0.08重量份),在15℃下反应1小时,使得式C中的C4上亚甲氨基还原成甲氨基,将反应后得到物料的pH值调整为6-7,用水萃取,除去水后得到萃取有机相物料,得到式D阿维菌素C4-2-硝基苯乙酰基,C5-甲氨基的氨化还原产物。
(4)将步骤(3)得到的式D所示物质溶于300mL无水二氯甲烷,溶解充分后,降温为15℃,加入锌粉8.5g(0.130摩尔),分批次加入氯化铵18.5g(0.136摩尔),控温为20℃反应5小时,使得式D中的C5上的2-硝基苯乙酰基脱除为羟基,得到式E阿维菌素C4-羟基,C5-甲氨基产物。
(5)向步骤(4)反应后的物料中加入等体积的水,水洗两次,水洗后的物料用无水硫酸钠进行干燥,得到式E物质的二氯甲烷溶液。向上述得到的式E物质的二氯甲烷溶液中加入14.0g苯甲酸(0.115摩尔),25℃下搅拌30min后,减压蒸馏去除二氯甲烷,即得甲胺基阿维菌素苯甲酸盐化合物,即甲维盐。
产品定性定量检测:
从步骤(1)中取出1mL物料(如式B所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图1所示,核磁碳谱如图2所示。从质谱图中可以发现m/z为1067.5670处的信号峰为[M+NH4]+的特征峰,从碳谱图中可以发现δ40.482,74.905,125.276,127.760,128.616,129.618,135.134,148.681,173.334处为2-硝基苯乙酰基环上碳原子的信号峰,由此证明式B中的C4-羟基中的氢替换为-2-硝基苯乙酰基。
从步骤(2)中取出1mL物料(如式C所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图3所示,核磁碳谱如图4所示。从质谱图中可以发现m/z为1065.5514处的信号峰为[M+NH4]+的特征峰,从碳谱图中可以发现δ40.459,74.888,125.251,127.724,128.601,129.589,135.047,148.656,173.310处为2-硝基苯乙酰基环上碳原子的信号峰,由此证明式C中的C4-羟基中的氢替换为-2-硝基苯乙酰基。从碳谱上可以发现δ205.869处为C5-羟基氧化成羰基上碳原子的信号峰,由此证明式C阿维菌素C5-羰基中的氢被氧化成羰基。
从步骤(3)中取出1mL物料(如式D所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图5所示,核磁氢谱如图6所示。从质谱图中可以发现m/z为1063.5704处的信号峰为[M+H]+的特征峰,从氢谱图中可以发现δ2.6076处的3个H为甲氨基上的H。由此证明式D阿维菌素C5-羰基被氨化还原成甲氨基。
从步骤(4)中取出1mL物料(如式E所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)分析,质谱图如图7所示。从质谱图中可以发现m/z为886.5301处的信号峰为[M+H]+的特征峰,由此证明式E中的C4上的2-硝基苯乙酰基脱除为羟基。
将步骤(5)得到产品中的甲维盐用核磁共振检测氢谱和碳谱,氢谱图如图8所示,碳谱图如图9所示。通过与甲维盐标准品的图谱进行比对,证明步骤(5)得到的产品确实为甲维盐。
将步骤(5)得到的产品中的甲维盐用HPLC进行定量分析,HPLC图如图10所示。124.36g甲维盐产品中甲维盐的纯度为84.5wt%,相对于步骤1中100g(0.115摩尔)的阿维菌素的投料量,甲维盐的产量为105.08g(0.104摩尔),由此计算收率为90.97%(相对于完全转化的理论产量)。
实施例2
(1)向1000mL三口瓶中加入100g(0.115摩尔)阿维菌素(购自湖北远成赛创科技有限公司,如式F所示的物质,阿维菌素B1a和阿维菌素B1b的重量比为1:20)和300mL无水二氯甲烷,溶解充分后,降温到-15℃,加入有机碱(四甲基乙二胺),降温到-20℃,缓慢滴加酰化试剂2-硝基苯乙酰氯28g(0.235摩尔)与无水二氯甲烷50ml的混合溶液,在-25℃下反应2小时,使得式F中的C4-羟基中的氢替换为2-硝基苯乙酰基,反应完成后,将反应后物料的pH值调整为6-7,用水萃取,除水后减压蒸馏,得到式B阿维菌素C4-2-硝基苯乙酰基,C5-羟基的单保护产物物料。
(2)将步骤(1)得到的单保护产物物料降温到-20℃,加入复合氧化试剂(二甲基亚砜、四甲基乙二胺和磷酸苯酯二酰氯组成)70g。在-25℃下反应2小时,使得式B中的C5-羟基氧化成羰基;将反应后得到物料的pH值调整为6-7,用水萃取,除去水后得到萃取有机相物料,得到式C阿维菌素C4-2-硝基苯乙酰基,C5-羰基的氧化产物物料。
(3)将步骤(2)得到的氧化产物物料升温到5℃,加入氨化试剂Ⅰ七甲基二硅氮烷110g(0.627摩尔)和催化剂Ⅱ氯化锌14g(0.14重量份),在5℃下反应4小时,使得式C中的C4上的羰基氨化为亚甲氨基。加入试剂Ⅲ甲醇150g(1.5重量份)和氨化还原剂Ⅳ硼氢化钠10g(0.10重量份),在10℃下反应2小时,使得式C中的C4上亚甲氨基还原成甲氨基,将反应后得到物料的pH值调整为6-7,用水萃取,除去水后得到萃取有机相物料,得到式D阿维菌素C4-2-硝基苯乙酰基,C5-甲氨基的氨化还原产物物料。
(4)将步骤(3)得到的氨化还原产物物料升温为到15℃,加入锌粉10g(0.153摩尔),分批次加入氯化铵20g(0.146摩尔),控温为25℃反应4小时,使得式D中的C4上的2-硝基苯乙酰基脱除为羟基,得到式E阿维菌素C4-羟基,C5-甲氨基产物物料。
(5)向步骤(4)反应后的物料中加入等体积的水,水洗两次,水洗后的物料用无水硫酸钠进行干燥,得到式E物质的二氯甲烷溶液。向上述得到的式E物质的二氯甲烷溶液中加入14.0g苯甲酸(0.115摩尔),25℃下搅拌30min后,减压蒸馏去除二氯甲烷,即得甲胺基阿维菌素苯甲酸盐化合物,即甲维盐。
产品定性定量检测:
从步骤(1)中取出1mL物料(如式B所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图1所示,核磁碳谱如图2所示。从质谱图中可以发现m/z为1067.5670处的信号峰为[M+NH4]+的特征峰,从碳谱图中可以发现δ40.482,74.905,125.276,127.760,128.616,129.618,135.134,148.681,173.334处为2-硝基苯乙酰基环上碳原子的信号峰,由此证明式B中的C4-羟基中的氢替换为-2-硝基苯乙酰基。
从步骤(2)中取出1mL物料(如式C所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图3所示,核磁碳谱如图4所示。从质谱图中可以发现m/z为1065.5514处的信号峰为[M+NH4]+的特征峰,从碳谱图中可以发现δ40.459,74.888,125.251,127.724,128.601,129.589,135.047,148.656,173.310处为2-硝基苯乙酰基环上碳原子的信号峰,由此证明式C中的C4-羟基中的氢替换为-2-硝基苯乙酰基。从碳谱上可以发现δ205.869处为C5-羟基氧化成羰基上碳原子的信号峰,由此证明式C阿维菌素C5-羰基中的氢被氧化成羰基。
从步骤(3)中取出1mL物料(如式D所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)和核磁共振进行分析,质谱图如图5所示,核磁氢谱如图6所示,从质谱图中可以发现m/z为1063.5704处的信号峰为[M+H]+的特征峰,从氢谱图中可以发现δ2.6076处的3个H为甲氨基上的H。由此证明式D阿维菌素C5-羰基被氨化还原成甲氨基。
从步骤(4)中取出1mL物料(如式E所示物质),萃取、减压蒸馏除去二氯甲烷后进行高分辨率质谱(HRMS)分析,质谱图如图7所示,从质谱图中可以发现m/z为886.5301处的信号峰为[M+H]+的特征峰,由此证明式E中的C4上的2-硝基苯乙酰基脱除为羟基。
将步骤(5)得到产品中的甲维盐用核磁共振检测氢谱和碳谱,氢谱图如图8所示,碳谱图如图9所示。通过与甲维盐标准品的图谱进行比对,证明步骤(5)得到的产品确实为甲维盐。
将步骤(5)得到的产品中的甲维盐用HPLC进行定量分析,HPLC图如图10所示,131.4g甲维盐产品中甲维盐的纯度为81.5重量%,相对于步骤1中100g(0.115摩尔)的阿维菌素的投料量,甲维盐的产量为107.09g(0.106摩尔),由此计算收率为92.71%(相对于完全转化的理论产量)。
以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种具有诱捕功能的甲氨基阿维菌素苯甲酸盐杀螨悬浮剂及其制备方法与流程
- 一种制备甲氨基阿维菌素苯甲酸盐的新晶型的方法及其应用与流程
- 纯化甲氨基阿维菌素‑苯甲酸盐的方法以及含有所述甲氨基阿维菌素‑苯甲酸盐的组合物与流程
- 一种甲氨基阿维菌素苯甲酸盐和溴虫腈的水分散粒剂及其制备方法和应用与流程
- 一种甲氨基阿维菌素苯甲酸盐和溴虫腈的水悬浮剂及其制备方法和应用与流程
- 复方乙酰氨基阿维菌素制剂及其制备方法和应用与流程
- 甲氨基阿维菌素苯甲酸盐泡腾片剂及其制备方法与流程
- 一种甲氨基阿维菌素B2苯甲酸盐的制备方法与流程
- 一种含有乙酰氨基阿维菌素的杀线剂组合物的制造方法与工艺
- 一种甲氨基阿维菌素苯甲酸盐和杀铃脲复配悬浮剂及其制备方法与流程
- 用于烯烃聚合的包含单酯和氨基苯甲酸酯内部供体的原催化剂的制造方法与工艺
- 一种苯甲酰甲酸甲酯的合成方法与制造工艺
- 一种对羟基苯甲酸乙酯的制备方法与制造工艺
- 一种邻乙氧基苯甲酰胺药物中间体邻乙氧基苯甲酸乙酯的合成方法与制造工艺
- 一种间溴苯甲酸的环保制备方法与制造工艺
- 一种喷托维林药物中间体1‑苯基环戊烷甲酸的合成方法与制造工艺
- 使用了2‑苯基‑4,4’‑二氨基二苯基醚类的清漆、成形性优异的酰亚胺树脂组合物及具有优异断裂伸长率的固化树脂成形体、以及使用了这些的预浸料、酰亚胺预浸料、及耐热性和机械强度优异的纤维强化材料的制造方法与工艺
- 氨基甲酸酯粘着剂的制造方法与工艺
- 一种抗肿瘤活性的高乌甲素靛红杂合体及其合成方法与制造工艺
- 一种具有抗肿瘤活性的高乌甲素氮杂肉桂酸杂合体及其合成方法与制造工艺