用于生产吩嗪‑1‑甲酰胺的基因工程菌株、制备方法及用途与流程
本发明涉及一种高产吩嗪-1-甲酰胺(pcn)的基因工程菌株、制备方法及其用途,属于基因工程技术领域。
背景技术:
到目前为止,科学家已经发现了超过6000种包含吩嗪结构的化合物,它们中绝大多数都是化学合成的,从生物体内分离得到的还不到100种。但是从生物体内分离到的吩嗪化合物多具有广谱的抗细菌、真菌等植物、动物病源菌的能力。例如,吩嗪-1-羧酸(pca)能够抑制引起小麦根部腐蚀的小麦全蚀病,吩嗪-1-甲酰胺则对西红柿枯萎病等病原菌有显著的抑制作用,而有些吩嗪衍生物则显示出了在医药领域的用途,已经有越来越多的学者正在研究其作用机理。天然吩嗪化合物多产自细菌,人工培养时在合适的条件下它们中多能大量合成并分泌吩嗪及其衍生物,产量在毫克每升到克每升的水平。这些优良性能都显示了吩嗪及其类似物大规模推广应用的巨大潜力。
假单胞菌中吩嗪的合成和调控基因均较为保守。早在1970年代,使用放射性同位素标记法已经发现吩嗪的生物合成途径与分枝酸(shikimicacid)途径有密切的关系(turnerjm,messengeraj.occurrence,biochemistryandphysiologyofphenazinepigmentproduction.advmicrobphysiol,1986,27:211-275)。通过对比不同吩嗪产生菌的合成序列发现,吩嗪的合成至少需要编码phza、phzd、phze、phzf和phzg蛋白的基因。多数吩嗪合成基因簇还包含一个编码3-脱氧-a-阿拉伯庚酮糖酸-7-磷酸(3-deoxy-d-arabino-heptulosonate-7-phosphate,dahp)合成酶phzc的基因,它可以催化分树酸途径的第一步,使dahp脱磷酸进而环化成苯环,经脱水、加氢反应形成莽草酸,进一步被修饰形成分支酸。phzc可促使初级代谢产物向吩嗪合成方向转化,其所催化的反应与芳香簇氨基酸合成途径类似。
整个吩嗪基因座除包含一套由7个基因组成的吩嗪合成基因外,还存在复杂的代谢调控网络。在假单胞菌中,吩嗪次级代谢产物的调控可分为转录调控(transcriptionalcontrol)和转录后调控(post-transcriptionalcontrol)两部分。比如群感应调节系统,在荧光与绿针假单胞菌中这两个基因被命名为phzi/phzr,并直接位于吩嗪合成基因的上游。中国专利zl200610023459.9通过改变铜绿假单胞菌m18中全局调控子gaca基因的表达,大幅提高了吩嗪-1-羧酸的产量。本课题申报的专利zl201310566864.5,通过敲除绿针假单胞菌ht66中吩嗪合成的调控基因psra或rpea基因,也显著提高了吩嗪-1-甲酰胺的产量。由于微生物细胞内的代谢调控网络复杂,不同调控基因相互作用,仅依靠改变调控基因,难以进一步提高次级代谢产物的产量。
本发明专利旨在通过敲除代谢途径的结构基因,改变细胞中相关物质的代谢流向,从而使中间代谢产物朝着吩嗪合成的方向积累,从而促进吩嗪类次级代谢产物的合成。
技术实现要素:
针对现有技术中的缺陷,本发明的目的是提供一种高产pcn的基因工程菌株及其制备方法和用途。
本发明是通过以下技术方案实现的:
第一方面,本发明提供了一种高产吩嗪-1-甲酰胺的基因工程菌株,是敲除了绿针假单胞菌(psedunomonaschlororaphis)ht66cctccno:m2013467及其衍生物基因组中的pyka基因,即得。
作为优选方案,所述pyka基因的碱基序列如seqidno.1所示。
作为优选方案,所述pyka基因的对应蛋白质的氨基酸序列如seqidno.2所示。
第二方面,本发明提供了一种如前述的高产吩嗪-1-甲酰胺的基因工程菌株的制备方法,包括插入突变法和无痕敲除法。
作为优选方案,所述插入突变法敲除pyka基因包括如下步骤:
s1、扩增pyka基因片段,并插入pex18tc质粒中;
s2、扩增卡那霉素或庆大霉素等抗性基因,插入pyka基因中间,使pyka基因发生插入突变;
s3、将突变后的pyka基因与ht66基因组中的pyka基因发生同源重组,根据抗性筛选出阳性克隆,即可。
作为优选方案,所述无痕敲除pyka基因法包括如下步骤:
a1、扩增pyka基因片段的上下游同源臂;
a2、融合pcr法连接上下游同源臂并插入pk18mobsacb质粒中;
a3、使重组质粒上的pyka基因的上下游同源臂片段与ht66基因组发生同源重组,利用蔗糖压力和抗性筛选出阳性克隆,即可。
第三方面,本发明还提供了一种用于培养前述的基因工程菌株的培养基,其包括如下组分:甘油、蛋白胨、酵母提取物、硫酸镁、磷酸氢二钾。
作为优选方案,所述的培养基包括按重量份数计的如下组分:
第四方面,本发明又提供了一种如前述的高产吩嗪-1-甲酰胺的基因工程菌株在生产吩嗪-1-甲酰胺中的用途。
其中,所述基因工程菌株在制备吩嗪-1-甲酰胺时的条件为:好氧培养;温度:28~32摄氏度;ph:6~8;转速200~350rpm。。
本发明的基本原理为:pyka是编码丙酮酸激酶的基因,在不同种属的微生物中相对比较保守。丙酮酸激酶是糖酵解途径中的一个关键酶,可以催化磷酸基团的转移将磷酸烯醇式丙酮酸(pep)转化成丙酮酸从而进入tca循环或其他途径。由于pep和丙酮酸处于细胞内糖、氨基酸和脂代谢的关键位置,且pep是莽草酸合成途径中的初始底物之一(赤藓糖-4-磷酸(e4p)是另一个底物),莽草酸途径的终产物分支酸是吩嗪-1-甲酰胺(pcn)合成的直接底物,故敲除pyka基因能够减弱pep转化成丙酮酸的反应,使更多的碳源流向莽草酸途径,从而提高pcn产量。
因此,与现有技术相比,本发明具有如下的有益效果:
采用全新的调控策略,改变细胞内代谢物质流向,大幅度提高了pcn的产量;该基因工程菌株安全可靠,稳定性好,可显著促进pcn的工农业应用。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为不同菌株中pyka基因扩增产物的电泳图(l1为空白对照,l2:以野生株ht66的基因组为模板,l3:以突变株ht66δpyka的基因组为模板);
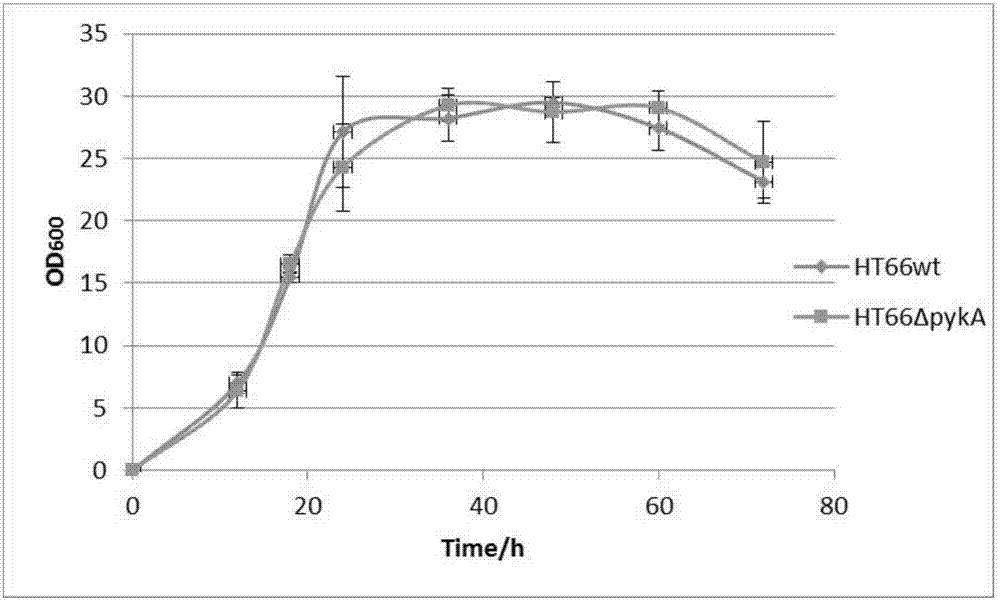
图2为野生株ht66与突变菌株ht66δpyka的生长曲线对比图;
图3为不同菌株中pyka基因扩增产物的电泳图(l1为空白对照,l2:以菌株p3的基因组为模板,l3:以突变株p3δpyka的基因组为模板)
图4为菌株p3和突变株p3δpyka的生长曲线对比图;
图5为野生株ht66和突变株ht66δpyka的pcn发酵曲线;
图6为菌株p3和突变菌株p3δpyka的pcn发酵曲线。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
本发明的绿针假单胞菌(psedunomonaschlororaphis)ht66,该菌株已在中国典型培养物保藏中心(简称cctcc)保藏,保藏单位地址为:中国.武汉.武汉大学,邮编为:430072,保藏日期为:2013年10月12日,保藏编号为:cctccno∶m2013467。菌株p3为ht66经过多重突变后获得的pcn高产菌株【张平原,彭华松,沈雪梅,等.高产吩嗪-1-甲酰胺的绿针假单胞菌的诱变与基因工程育种.上海交通大学学报:农业科学版,2015,33(2):90-94.】。
实施例1
本实施例涉及一种以ht66基因组为模板,利用插入突变法制备ht66δpyka基因工程菌株的方法,包括如下步骤:
1.引物设计
以pyka基因序列为参考,利用primer5设计引物并合成,引物序列如下,下划线表示限制性内切酶hindiii和saci的酶切位点。
pyka1:ccaagcttatgtccgtccgtcgtaccaa(seqidno.3)
pyka2:acgagctctcagaccattgggtcgcca(seqidno.4)
以质粒pbbb5k为模板,设计卡那抗性基因(kan)的引物并合成,引物序列如下,下划线表示限制性内切酶xhoi的酶切位点。
pbb-kan-f:ccctcgagggaattgccagctggggcgcseqidno.5
pbb-kan-r:ccctcgagtcagaagaactcgtcaagaagseqidno.6
2.插入突变重组质粒构建
以ht66基因组和pbbb5k质粒为模板,利用pyka1/2和pbb-kan-f/r引物通过pcr分别扩增pyka基因(1452bp)和kan基因(1000bp),扩增产物纯化后,kan基因pcr产物先置于-20°冰箱保存。将pyka基因和pex18tc质粒利用hindiii和saci分别进行双酶切,酶切体系切胶回收,16℃连接并转化e.colidh5a感受态细胞,通过tc(20ppm)抗性平板筛选获得阳性克隆。提取pex-pyka质粒,与kan基因一起分别进行xhoi单酶切,割胶回收后16℃连接过夜并转化dh5a,使用含tc(20ppm),kan(50ppm)的抗性平板筛选重组子获得阳性克隆,提取获得的质粒pex-pyka-kan,转入e.colism10感受态细胞。
3.插入突变菌株筛选
将转入pex-pyka-kan质粒的sm10与菌株ht66进行双亲杂交,通过tc(150ppm),kan(50ppm),sp(100ppm)的lb抗性平板进行单交换筛选,含15%蔗糖和kan(50ppm),sp(100ppm)的lb平板进行双交换筛选,外部引物(pyka1/2)pcr验证,提基因组扩增送测序,最后获得插入突变株ht66δpyka。
利用pyka1和pyka2引物进行pcr验证,插入突变菌株可扩增出长约2500bp的片段(pyka和kan基因大小之和),而未敲除的野生菌株则可扩增出长约1500bp的片段(pyka大小为1452bp),结果如图1所示。图1中:m为marker,l1为空白对照,l2:以菌株ht66的基因组为模板,l3:以菌株ht66δpyka的基因组为模板。
通过液体培养,测定菌株ht66和ht66δpyka生长曲线如图2所示。结果显示在ht66中敲除pyka基因不会影响其生长,表明该基因工程菌的稳定性好,有利于对该菌进行进一步改造。
实施例2
本实施例涉及一种高产pcn的基因工程菌株的制备,出发菌株p3是绿针假单胞菌ht66经过多重突变后获得的高产pcn的菌株。具体包括如下步骤:
1、引物设计:
以pyka基因上下游序列为参考,利用primer5设计敲除引物并合成,引物序列如下(下划线表示xbai和hindiii酶切位点):
pykaf1:ccggggatcctctagaaagatcgttacaacgcggtcg(seqidno.7)
pykar1:tggtacgacggacggacatg(seqidno.8)
pykaf2:tgtccgtccgtcgtaccaacccaatggtctgagccacc(seqidno.9)
pykar2:ggccagtgccaagcttcgagttcggttccagcctg(seqidno.10)
2、重组质粒构建:
以p3基因组为模板,利用pykaf1、pykar1和pykaf2、pykar2通过pcr扩增pyka上下游同源臂片段,pcr产物纯化后,利用in-fusionhdcloningkit方法将该上下游同源臂连接至已进行酶切过的pk18mobsacb质粒上并转化e.colidh5a,通过蓝白斑筛选,白斑菌落利用引物pykaf1和pykar2进行pcr验证后送测序。
3、突变菌株筛选:
将测序正确的质粒pk18-pyka转化至e.colis17,与菌株p3进行结合转移,通过kan和amp抗性平板单交换筛选,15%(w/v)蔗糖板双交换筛选,外部引物pcr验证,提基因组扩增送测序,最后获得菌株p3的pyka敲除突变株,命名为p3δpyka,利用pykaf1和pykar2引物pcr扩增,敲除菌株可扩增出长约1100bp的pyka上下游同源臂片段,而未敲除的野生菌株则可扩增出长约2500bp的片段(pyka大小为1452bp),表明pyka基因成功敲除,结果如图3所示。图3中,l1为空白对照,l2:以菌株p3的基因组为模板,l3:以突变株p3δpyka的基因组为模板。
通过在kb培养基进行液体培养,同时测定菌株p3和p3δpyka生长曲线,如图4所示。结果表明,在菌株p3中敲除pyka基因不会影响其生长,表明此基因工程菌株稳定性非常好,有利于其工业化应用。
实施例3
本实施例涉及一种利用实施例1制备的基因工程菌株合成pcn的方法,包括如下步骤:
分别将菌株ht66δpyka和ht66wt(野生株ht66作为对照)分别在kb固体平板上活化两次,然后接至5mlkb液体培养基培养,培养至对数期,以2%的比例转接至装有60mlkb培养基的三角瓶进行发酵。发酵条件为28℃,180rpm。定时取样,以hplc测定菌株pcn产量。
hplc检测条件:
流动相为乙腈-25mm乙酸铵,色谱柱为wondasilc18-wrreversephasecolumn(5μm;4.6×250mm,shimadzu,japan),检测波长254nm,检测条件:0-2min,8%乙腈-92%25mm乙酸铵,2-20min,乙腈浓度从8%升至60%,乙酸铵浓度从92%下降至40%,20-21min,8%乙腈-92%25mm乙酸铵。
分别测定菌株ht66和菌株ht66δpyka的pcn产量,结果如图5所示。从图可见,敲除pyka基因可以明显提高pcn产量,对照株ht66的pcn产量为552mg/l,而突变株在48h时其pcn产量为1038mg/l,相比提高了88%。由此可见,在绿针假单胞菌ht66中敲除pyka基因能够有效改变物质的流向,有利于积累pcn。
实施例4
本实施例涉及一种利用实施例2制备的基因工程菌株生产pcn的方法,包括如下步骤:
首先将菌株p3δpyka和p3(对照)分别在kb固体平板上活化,然后将其接至5mlkb液体培养基培养,待其生长到对数期后分别接种至装有60mlkb培养基的三角瓶进行发酵。初始接种量od600为0.02。发酵条件为28℃,180rpm。定时取样,分别测定菌株p3δpyka和p3发酵液中的pcn产量,结果如图6所示。
从图可见,敲除pyka基因可以明显提高pcn产量,在36h时其pcn产量为3594mg/l,与同样条件下对照株p3的pcn产量(1923mg/l)相比,提高了71.3%。该结果表明,在绿针假单胞菌p3中敲除pyka基因能够有效的再分配细胞碳、氮源的流向,使其流向莽草酸途径,进而流向pcn合成。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 大豆低温诱导人工合成启动子SP5及应用的制造方法与工艺
- 一种来源于虎眼万年青的尿苷-5’-二磷酸半乳糖差向异构酶,其核苷酸序列及应用的制造方法与工艺
- 一种来源于虎眼万年青的尿苷-5’-二磷酸木糖差向异构酶,其核苷酸序列及应用的制造方法与工艺
- 新的赖氨酸脱羧酶突变体及其应用的制造方法与工艺
- 一种嗜热中性蛋白酶基因、工程菌、酶及其应用的制造方法与工艺
- 一种碱性耐盐普鲁兰酶PulA及其基因和应用的制造方法与工艺
- 一种热稳定性提高的β‑淀粉酶突变体的制造方法与工艺
- 一类耐热型植酸酶突变体及其编码基因和应用的制造方法与工艺
- 含CCL5和SSTR2基因的重组痘病毒及其制备方法与流程
- 一种催化黄酮类化合物葡萄糖苷化的基因工程菌及其应用的制造方法与工艺
- 一种基因组DNA提取方法与流程
- 一种生产3‑羟基丙酸共聚物的重组菌及其构建方法和应用与流程
- 半胱氨酸双加氧酶、基因及其催化产物半胱亚磺酸的新用途的制造方法与工艺
- 一种用于鱼类基因工程实验的便于捕鱼的鱼缸的制造方法与工艺
- 一种利用CRISPR/Cas9在哺乳动物细胞中实现大片段DNA定点整合的方法与流程
- 一种AAV6型腺相关病毒体外高效感染T细胞的方法与流程
- 一种高效籼稻遗传转化方法与流程
- 在基因打靶中具有高突变效率的PL‑LbCpf1‑RR基因及其应用的制造方法与工艺
- ZmRCI2‑8 基因及其在促进非生物胁迫条件下植物发芽和侧根生长中的应用的制造方法与工艺
- 一种水稻种子特异性启动子Posseed及其应用的制造方法与工艺