一种多孔氮掺杂碳电极材料及其制备方法与流程
本发明涉及一种多孔氮掺杂碳电极材料及其制备方法。属于超级电容器电极材料制备领域,
背景技术:
超级电容器作为一种新型的储能装置,以其高功率密度、长使用寿命和快充放电速度等优点被广泛用于混合电动汽车和便携式电子设备,已成为近年来的研究热点。
活性炭、碳纳米管和碳气凝胶等碳材料是超级电容器中常见的电极材料,具有良好的导电性和稳定性等优点。但是将碳材料组装成超级电容器时,其比电容值较小,从而限制其工业发展。通过向碳材料表面引入官能团或杂原子(o、n、b、s)是提高电极材料电容值的一个有效的方法。其中氮是碳材料掺杂的理想元素,通过氮掺杂可以有效地改善碳纳米材料的力学、电化学、催化、吸附等性能。高聚物进行热解处理是制备氮掺杂碳材料的有效方法之一。其中,聚苯胺因具有含氮丰富、形貌可控、价格低廉而成为理想的氮源。
目前,申请号为cn201810017931.0公布了一种氮磷硫共掺杂的介孔碳球及其制备方法,利用聚苯胺掺杂有机酸-植酸为碳前驱体,纳米二氧化硅溶胶为致孔剂,制备得到氮、磷、硫三种杂原子共掺杂的碳材料。申请号为cn201610548972.3提供了一种二氧化硅修饰的多球腔碳材料的制法,首先以正硅酸乙酯为原料制备得到二氧化硅小球,以该二氧化硅小球为致孔剂制备得到二氧化硅/聚苯胺复合材料。在所报道的多孔杂原子掺杂碳材料的制备方法中,二氧化硅溶胶、sba-15等是常用的致孔剂。然而,这些致孔剂材料价格较为昂贵,制备条件较为苛刻,在实际的应用中受到了一定的限制。本发明主要是将经济成本低廉的纳米二氧化硅作为致孔剂,在二氧化硅、质子酸、苯胺分子、聚苯胺大分子间的氢键作用力和静电力的双重组装作用下,制备得到多孔氮掺杂碳电极材料,并获得了较好的电化学效果。
技术实现要素:
本发明的目的在于提供一种用于超级电容器的多孔氮掺杂碳电极材料及其制备方法。多孔氮掺杂碳电极材料为氮元素掺杂和多孔结构的特殊复合材料。多孔结构提高了电极材料与电解质的接触面积,利于离子的快速交换。氮元素掺杂后,所得电极材料的能量储存功能得到极大提高。
为了实现上述目的,本发明所述的多孔氮掺杂碳电极材料通过以下步骤获得:
(1)致孔剂/氮源材料复合体的制备;
(2)氮掺杂碳材料的制备;
(3)多孔氮掺杂碳材料的制备。
步骤(1)所述的致孔剂是指不同粒径的二氧化硅及其衍生物。
步骤(1)所述的氮源材料是指含氮元素的聚苯胺、聚吡咯及其衍生物。
步骤(1)所述的致孔剂/氮源材料复合体的制备是指将致孔剂分散在0.05~2moll-1的酸性水溶液中,超声分散0.5~2h,其浓度为5~25wt%,超声频率为30~50khz,所述酸性水溶液为盐酸、硫酸、高氯酸或十二烷基苯磺酸的水溶液;在上述混合溶液中加入氮源材料的前驱体,混合溶液的温度控制在0~4℃,在一种新型的搅拌方式下分散0.5~2h,氮源材料的前驱体的浓度为0.05~0.5moll-1,氮源材料的前驱体可以是苯胺、吡咯及其衍生物;将氧化剂分散在0.05~2moll-1的酸性水溶液中,0~4℃下冷藏0.5~2h,氧化剂的浓度为0.05~0.5moll-1,所述氧化剂为过硫酸铵、过硫酸钾和过氧化氢;在一种新型的搅拌方式下,在含氮源材料的前驱体的混合液中逐滴加入氧化剂混合溶液,保持温度为0~4℃;待氧化剂滴加完毕后,在一种新型的搅拌方式下,反应时间为0.5~2h。反应结束后,将混合液依次用水和无水乙醇洗至滤液无色,真空干燥后得到致孔剂/氮源材料复合体。
步骤(1)所述的一种新型的搅拌方式为机械搅拌和超声波并用的分散混合方式,机械搅拌转速为200~400rpm,超声波频率为30~50khz。
步骤(2)所述的氮掺杂碳材料的制备是指在惰性气体的保护下,将步骤(1)所述的致孔剂/氮源材料复合体放入管式马弗炉中,在700~1000℃下进行碳化3~8h,冷却后得到氮掺杂碳材料。
步骤(2)所述的惰性气体为氮气或氩气。
步骤(3)所述的多孔氮掺杂碳材料的制备是指将步骤(2)中所述的氮掺杂碳材料置于刻蚀溶液中浸泡30min~7h,除去致孔剂,经离心分离、真空干燥得到。
步骤(3)所述的刻蚀溶液是指5~10wt%的氢氟酸溶液或0.25~1moll-1的热氢氧化钠水溶液。
步骤(3)所述的热氢氧化钠水溶液是指温度为70~90℃的溶液。
本发明的优点在于:本发明所用致孔剂具有来源广泛、价格低廉和工艺成熟等优点。本发明所用的机械搅拌和超声波并用的分散方式具有分散效果显著,提高反应效率和有效避免纳米颗粒的团聚现象等优点。本发明将氮源材料通过高温碳化得到氮掺杂碳材料具有操作简便,产率高等优点,可实现工业化。本发明的产品之所以可以工业化,是因为可以根据实际生产需要,在一定条件下任意加大原料(二氧化硅等)的量从而实现工业化生产。本发明得到的多孔氮掺杂碳材料具有工艺简单、成本低廉、导电性良好、循环稳定性好、热稳定性优异等优点,可作为电极材料应用到超级电容器中,具有良好的工业应用前景。
附图说明
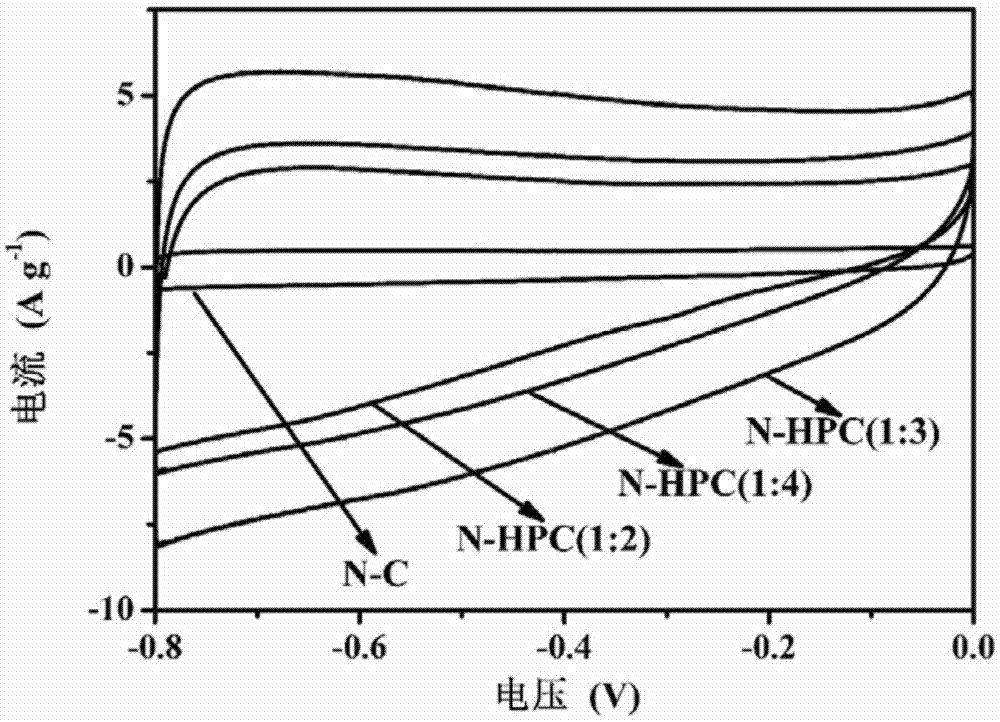
图1为实施例1和实施例2氮掺杂碳材料在6m氢氧化钾水溶液中50mvs-1扫描速度下的循环伏安曲线,其中横坐标为电位,单位为v;纵坐标为电流,单位为ag-1。
图2为实施例1和实施例2氮掺杂碳材料在不同电流密度下的比电容值,其中横坐标为电流密度,单位为ag-1;纵坐标为比电容值,单位为fg-1。
图3为实施例1中多孔氮掺杂碳材料的循环寿命测试曲线,其中横坐标为循环圈数;左侧纵坐标为比电容值,单位为fg-1;右侧纵坐标为电容保持率,单位为%。
图4为实施例1中pani/sio2(1:3)复合材料的扫描电镜图。
图5为实施例1中n-hpc(1:3)电极材料的扫描电镜图。
具体实施方式
实施例1:
(1)在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下,将平均粒径为20nm的二氧化硅分散在70ml1moll-1的盐酸水溶液中2h。在上述混合溶液中加入苯胺,混合溶液的温度控制在0℃,在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下混合0.5h,苯胺的浓度为0.5moll-1(苯胺与二氧化硅的质量比分别为1:2,1:3和1:4);将过硫酸铵分散在1moll-1的盐酸水溶液中,0℃下冷藏0.5h,过硫酸铵的浓度为0.5mol/l。在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,在含苯胺的混合液中逐滴加入过硫酸铵的混合溶液,保持温度为0℃;待过硫酸铵混合液滴加完毕后,在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,控制反应温度0℃,反应12h后结束,将混合液依次用水和无水乙醇洗至滤液无色,真空干燥后得到致孔剂/氮源材料复合体,命名为pani/sio2(1:2)、pani/sio2(1:3)和pani/sio2(1:4)。
(2)将致孔剂/氮源材料复合体在氮气的保护下,在800℃下进行碳化2h,冷却后得到氮掺杂碳材料,命名为n-c/sio2(1:2)、n-c/sio2(1:3)和n-c/sio2(1:4)。
(3)将氮掺杂碳材料置于20wt%的氢氟酸溶液中浸泡24h,离心分离,真空干燥得到多孔氮掺杂碳材料,命名为n-hpc(1:2)、n-hpc(1:3)和n-hpc(1:4)。
图4所示为pani/sio2(1:3)的扫描电镜图。在机械搅拌、超声分散作用下,二氧化硅纳米粒子均匀地覆盖在聚苯胺短纤维表面,得到表面形貌较规则的复合材料,说明二氧化硅的团聚现象得到改善。
图5为n-hpc(1:3)的扫描电镜图。经碳化、刻蚀后,电极材料的表面形貌为多孔的短纤维。
实施例2:
(1)在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下,将苯胺在70ml1moll-1的盐酸水溶液中2h,混合溶液的温度控制在0℃,在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下混合0.5h,苯胺的浓度为0.5moll-1;将过硫酸铵分散在1moll-1的盐酸水溶液中,0℃下冷藏0.5h,过硫酸铵的浓度为0.5moll-1。在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,在含苯胺的混合液中逐滴加入过硫酸铵的混合溶液,保持温度为0℃;待过硫酸铵混合液滴加完毕后,在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,控制反应温度0℃,反应12h后结束,将混合液依次用水和无水乙醇洗至滤液无色,真空干燥后得到聚苯胺pani。
(2)将聚苯胺在氮气的保护下,在800℃下进行碳化2h,冷却后得到氮掺杂碳材料,命名为n-c。
实施例3:
(1)在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下,将平均粒径为20nm的二氧化硅分散在70ml1moll-1的盐酸水溶液中2h。在上述混合溶液中加入苯胺,混合溶液的温度控制在0℃,在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下混合0.5h,苯胺的浓度为0.5moll-1(苯胺与二氧化硅的质量比分别为1:3);将过硫酸铵分散在1moll-1的盐酸水溶液中,0℃下冷藏0.5h,过硫酸铵的浓度为0.5moll-1。在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,在含苯胺的混合液中逐滴加入过硫酸铵的混合溶液,保持温度为0℃;待过硫酸铵混合液滴加完毕后,在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,控制反应温度0℃,反应12h后结束,将混合液依次用水和无水乙醇洗至滤液无色,真空干燥后得到致孔剂/氮源材料复合体,命名为pani/sio2(1:3)。
(2)将致孔剂/氮源材料复合体在氮气的保护下,分别在700、900℃下进行碳化2h,冷却后得到氮掺杂碳材料,命名为n-c/sio2(1:3)-700、n-c/sio2(1:3)-900。
(3)将氮掺杂碳材料置于20wt%的氢氟酸溶液中浸泡24h,离心分离,真空干燥得到多孔氮掺杂碳材料,命名为n-hpc(1:3)-700、n-hpc(1:3)-900。
实施例4:
(1)在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下,将平均粒径分别为7、40nm的纳米二氧化硅分散在70ml1moll-1的盐酸水溶液中2h。在上述混合溶液中加入苯胺,混合溶液的温度控制在0℃,在机械搅拌转速为300rpm和超声波频率为40khz并用的分散方式下混合0.5h,苯胺的浓度为0.5moll-1(苯胺与二氧化硅的质量比分别为1:3);将过硫酸铵分散在1moll-1的盐酸水溶液中,0℃下冷藏0.5h,过硫酸铵的浓度为0.5moll-1。在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,在含苯胺的混合液中逐滴加入过硫酸铵的混合溶液,保持温度为0℃;待过硫酸铵混合液滴加完毕后,在机械搅拌转速为300rpm和超声波频率为40khz的分散方式下,控制反应温度0℃,反应12h后结束,将混合液依次用水和无水乙醇洗至滤液无色,真空干燥后得到致孔剂/氮源材料复合体,命名为pani/sio2(1:3)-7、pani/sio2(1:3)-40。
(2)将致孔剂/氮源材料复合体在氮气的保护下,在800℃下进行碳化2h,冷却后得到氮掺杂碳材料,命名为n-c/sio2(1:3)-7、n-c/sio2(1:3)-40。
(3)将氮掺杂碳材料置于20wt%的氢氟酸溶液中浸泡24h,离心分离,真空干燥得到多孔氮掺杂碳材料,命名为n-hpc(1:3)-7、n-hpc(1:3)-40。
实施例5:
其他条件同实施例1,只是搅拌方式分别为:
a在机械搅拌转速为150rpm和超声波频率为60khz并用的分散方式下;
b在机械搅拌转速为500rpm和超声波频率为20khz并用的分散方式下;
c在机械搅拌转速为200rpm和超声波频率为60khz并用的分散方式下;
d在机械搅拌转速为450rpm和超声波频率为30khz并用的分散方式下。
上述四种搅拌方式得到致孔剂/氮源材料复合体团聚现象严重,无法进行下一步实验。
实施例6:
多孔氮掺杂碳材料的循环伏安测试:将实施例1和实施例2所得的氮掺杂碳材料配置成浓度为1mg/ml的水溶液,超声分散后待用。
选用三电极测试方法,以玻碳电极、铂丝和甘汞电极分别作为工作电极、对电极和参比电极。选用6m氢氧化钾水溶液作为电解液。循环伏安测试的电压窗口为-0.8-0v,扫描速率为50mvs-1。测试结果见图2。
多孔氮掺杂碳材料的电化学性能可以通过调节二氧化硅致孔剂的用量来实现。从图2的循环伏安图可以看出,n-c电极材料在50mvs-1的扫描速度,曲线覆盖面积最小,n-hpc(1:3)电极材料的曲线覆盖面积最大,可见,随着二氧化硅致孔剂的用量增加,材料中多孔的结构逐渐增多,利用电荷的快速移动,利于材料的电化学性能提高。但是,二氧化硅致孔剂用量过多时,材料的电化学性能降低,可能由于二氧化硅的团聚现象所致。因二氧化硅团聚后,会形成较大粒径的二氧化硅聚集体,影响电极材料的多孔结构。
实施例7:
多孔氮掺杂碳材料的充放电测试:将实施例1和实施例2所得的氮掺杂碳材料配置成浓度为1mg/ml的水溶液,超声分散后待用。
选用三电极测试方法,以玻碳电极、铂丝和甘汞电极分别作为工作电极、对电极和参比电极。选用6m氢氧化钾水溶液作为电解液。充放电测试的电压窗口为-0.8-0v,电流密度分别为0.5,1,2,5和10ag-1。测试结果见图3。
四种电极材料的充放电测试结果与循环伏安测试结果一致,其中,n-hpc(1:3)电极材料在电流密度为0.5ag-1时,其比电容可达218.75fg-1,10ag-1电流密度下,其比电容为117.5fg-1,电容保持率达53.7%,表现出良好的倍率特性。
实施例8:
多孔氮掺杂碳电极材料的循环寿命测试:将实施例1中所得的n-hpc(1:3)配置成浓度为1mg/ml的水溶液,超声分散后待用。
选用三电极测试方法,以玻碳电极、铂丝和甘汞电极分别作为工作电极、对电极和参比电极。选用6m氢氧化钾水溶液作为电解液。循环寿命测试方法同充放电测试,其电压窗口为-0.8-0v,电流密度为10ag-1,充放电次数为1000圈。测试结果见图4。
n-hpc(1:3)电极材料经1000次充放电测试后,其比电容保持率高达99%,表明材料具有优异的使用寿命。
以上所述仅为本发明的交叉实例而已,并不用以限制本发明,对本领域技术人员来说很显然可以做很多的改进,凡在本发明的精神和原则之内,所做的任何修改、同等替换、改进等,均应包含在本发明保护的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!