趋化因子受体拮抗体及其联合疗法的制作方法
本发明属于药物领域。具体而言,本发明涉及作为趋化因子受体拮抗剂的肽在栓塞疗法期间用于治疗对象的癌症或者降低或停止肿瘤生长中的应用。
背景技术:
细胞因子是由调节免疫应答的各种细胞包括单核细胞或淋巴细胞分泌的可溶性蛋白质。趋化因子是趋化蛋白质的超家族。趋化因子调节各种生物应答并且它们促进多个白细胞和淋巴细胞谱系募集到身体器官组织。趋化因子可以根据蛋白质中前两个半胱氨酸残基的相对位置分类为两个家族。在一个家族中,所述前两个半胱氨酸被一个氨基酸残基隔开,例如,CXC趋化因子,而在另一个家族中,所述前两个半胱氨酸是相邻的,例如CC趋化因子。
趋化因子的分子靶标是细胞表面受体。一种这样的受体是CXC趋化因子受体4(CXCR4),其是七次跨膜蛋白质,与G1偶联,并且以前称为LESTR(Loetscher,M.,Geiser,T.,O'Reilly,T.,Zwahlen,R.,Baggionlini,M.,和Moser,B.,(1994)J.Biol.Chem,269,232-237)、HUMSTR(Federsppiel,B.,Duncan,A.M.V.,Delaney,A.,Schappert,K.,Clark-Lewis,I.,和Jirik,F.R.(1993)Genomics 16,707-712)和融合素(Feng,Y.,Broeder,C.C,Kennedy,P.E.,和Berger,E.A.(1996)HIV-1进入辅因子:七次跨膜G蛋白偶联受体的功能性cDNA克隆(HIV-1entry cofactor:Functional cDNA cloning of a seven-transmembrane G protein-coupled receptor),Science 272,872-877)。CXCR4在造血来源的细胞上广泛表达,并且对于人类免疫缺陷性病毒1(HIV-1)而言,是CD4+的主要辅助受体(Feng,Y.,Broeder,C.C,Kennedy,P.E.,和Berger,E.A.(1996)HIV-1进入辅因子:七次跨膜G蛋白偶联受体的功能性cDNA克隆,Science 272,872-877).
目前,唯一已知的天然CXCR4配体是基质细胞衍生因子1(SDF-1)。基质细胞衍生因子-1α(SDF-1α)和基质细胞衍生因子-1β(SDF-1β)是密切相关的成员(在本文中共同称作SDF-1)。SDF-1α和SDF-1β的天然氨基酸序列是已知的,编码这些蛋白质的基因组序列亦然(1996年10月8日颁发的美国专利No.5,563,048,和1998年5月26日颁发的美国专利No.5,756,084)。
SDF-1在功能上不同于其他趋化因子,因为据报道它在骨髓祖细胞的运输、输出和归巢中具有重要作用(Aiuti,A.,Webb,I.J.,Bleul,C,Springer,T.,和Guierrez-Ramos,J.C,(1996)J.Exp.Med.185,111-120,以及Nagasawa,T.,Hirota,S.,Tachibana,K.,Takakura N.,Nishikawa,S.-I.,Kitamura,Y.,Yoshida,N.,Kikutani,H.,和Kishimoto,T.,(1996)Nature 382,635-638)。SDF-1结构上也不同,因为它与其他CXC趋化因子只有约22%的氨基酸序列同一性(Bleul,C.C,Fuhlbrigge,R.C,Casasnovas,J.M.,Aiuti,A.,和Springer,T.A.,(1996)J.Exp.Med.184,1 101-1109)。SDF-1看来是由几种细胞类型组成性产生的,并且在骨髓基质细胞中发现特别高的水平(Shirozu,M.,Nakano,T.,Inazawa,J.,Tashiro,K.,Tada,H.Shinohara,T.,和Honjo,T.,(1995)Genomics,28,495-500,以及Bleul,C.C,Fuhlbrigge,R.C,Casasnovas,J.M.,Aiuti,A.,和Springer,T.A.,(1996)J.Exp.Med.184,1101-1109)。SDF-1序列在物种之间的高度保守性暗示了SDF-1的基本生理作用。在体外,SDF-1刺激各种各样的细胞包括单核细胞和骨髓源性祖细胞的趋化性(Aiuti,A.,Webb,I.J.,Bleul,C,Springer,T.,和Guierrez-Ramos,J.C.,(1996)J.Exp.Med.185,111-120,以及Bleul,C.C,Fuhlbrigge,R.C,Casasnovas,J.M.,Aiuti,A.,和Springer,T.A.,(1996)J.Exp.Med.184,1101-1109)。特别值得注意的是它具有刺激高比例的静息和活化T-淋巴细胞的能力(Bleul,C.C,Fuhlbrigge,R.C,Casasnovas,J.M.,Aiuti,A.,和Springer,T.A.,(1996)J.Exp.Med.184,1101-1 109,以及Campbell,J.J.,Hendrick,J.,Zlotnik,A.,Siani,M.A.,Thompson,D.A.,和Butcher,E.C,(1998)Science,279 381-383)。
SDF-1的3-维晶体结构已被描述(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。SDF-1的结构活性分析(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)指出,虽然N-端残基1-8或1-9参与受体结合,但所述1-8和1-9肽单独没有展现出表明受体结合的体外活性,支持一则报道的结论,即所述肽没有采取与所述受体结合所必需的构象。该结果被认为暗示了蛋白质支架的其余部分、和/或所述蛋白质中其它地方的各种共有受体结合位点对于介导N-端与所述受体结合的构象要求而言是重要的(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。基于这些结果,已经对SDF-1与CXCR4结合提出了双位点模型,包括在残基1-17中的两个结合位点:N端位点和上游RFFESH位点(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。所述两个推定的结合位点以序列:KPVSLSYR-CPC-RFFESH为特征,其中所述两个推定的结合位点由整个CXC趋化因子家族特征性的CXC基序连接(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。这两个推定的结合区域已经在其他CC和CXC趋化因子中被鉴定为重要(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007,以及Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。这与下述的发现一致,即:虽然多种多样的趋化因子的N-端区域对于受体活化至关重要,但除了SDF-1以外的趋化因子的N端肽据报道缺乏受体结合活性并且不会是受体激动剂(Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007,以及Crump,M.,Gong J.-H.,Loetscher,P.,Rajarathnam,K.,Amara,A.,Arenzana-Seisdedos,F.,Virelizier,J.-L.,Baggiolini,M.,Sykes,B.D.,和Clark-Lewis,I.,(1997)EMBO J.,16,6996-7007)。
与CXCR4是HIV-1主要辅助受体的事实一致,SDF-1阻断HIV-1进入CD4+细胞中(Oberlin,E.,Amara,A.,Bachelerie,F.,Bessia,C,Virelizier,J.-L.,Arenzana-Seisdedos,F.,Schwartz,O.,Heard,J.-M.,Clark-Lewis,I.,Legler,D.F.,Loetscher,M.,Baggiolini,M.,和Moser,B.,(1996)Nature,382,833-835,以及Bleul,C.C,Farzan,M.,Choe,H.,Parolin,C,Clark-Lewis,I.,Sodroksi,J.,和Springer,T.A.,(1996)Nature,382,829-833)。已在努力鉴定选择性干扰HIV进入、但不干扰SDF-1信号传导的SDF-1衍生肽(Heveker,N.等,1998,Current Biology 8(7):369-376)。已提出了使用SDF-1的多种多样的潜在CXCR4结合片段来阻断HIV感染(WO 9728258,1997年8月7日公布;WO 9804698,1998年2月5日公布)。正如这些参考文献所澄清的,SDF-1、或SDF-1的片段的抗HIV活性不依赖对所述CXCR4受体的拮抗作用。
干扰素γ是由活化的T淋巴细胞(T细胞)释放的重要的细胞因子并且担任强效免疫调节剂。T细胞体内产生干扰素γ可导致身体内其他细胞释放能够调控免疫应答的许多方面的其他细胞因子、酶和抗体。影响活化的T细胞产生干扰素γ的能力的药剂被定性为免疫调节剂。
自身免疫疾病是通常据了解是由白细胞、特别包括T细胞过度产生细胞因子、淋巴毒素和抗体而引起的一组疾病。在自身免疫反应期间,T细胞据了解释放化学介质例如干扰素γ,所述化学介质导致自身免疫反应的病理症状的发展。自身免疫疾病的治疗因此包括使用能够抑制干扰素γ从T细胞释放的药剂。这样的自身免疫疾病可以包括,例如,多发性硬化(MS)、格林-巴利(Guillain-Barre)综合征、肌萎缩侧索硬化、帕金森氏(Parkinson's)病、阿尔茨海默氏(Alzheimer's)病、痛风、狼疮、和T细胞在其中发挥重要作用的任何其它人类疾病。
干扰素β是已经发现在治疗各种自身免疫疾病中具有治疗性应用的细胞因子。在自身免疫疾病例如MS中,Thl型T细胞的活化被认为是自身免疫应答的主要组成部分。在MS中,所述自身免疫应答攻击髓鞘神经元轴突。Thl细胞活化的经典标志之一是产生干扰素γ。在开发干扰素β作为治疗MS的治疗剂中,进行研究以证明干扰素β在体外降低淋巴细胞生产干扰素γ的速率的能力(Ann.Neurol.1998;44:27-34和Neurology 1998;50:1294-1300)。通过用干扰素β治疗减少干扰素γ释放指示了干扰素β在治疗MS中的有效性。对于抑制干扰素γ产生的其他药剂,特别是用于治疗自身免疫疾病的药剂,包括可以协同作用以提高现有药剂例如干扰素β的效应的药剂,一直有需求。
实体肿瘤生长通常是血管生成(新生血管化)依赖性的,而血管生成抑制剂因此已经被用作治疗实体肿瘤和转移的药剂。脉管系统中的内皮细胞(EC)在血管生成中发挥必要的作用,因此需要靶定这种活性的治疗剂。据了解,在血管生成期间血管内皮细胞的增殖、迁移和分化在正常和疾病状态二者中是由各种趋化因子和趋化因子受体的复杂的相互作用调控的。CXCR4在血管EC上表达,并且在这样的细胞中据报道是所有考查的趋化因子受体当中最丰富的受体(Gupta等,1998)。
现有技术(美国专利申请No.11/136,097)指出,CXCR4受体配体,具有类似于SEQ ID NO.1、SEQ ID NO.2或SEQ ID NO.3的氨基酸序列的SDF-1多肽拮抗剂,在各种实施方式中展现出抗血管生成和抗肿瘤活性。
技术实现要素:
在此,本发明人证明了在大鼠中这些SDF-1拮抗剂与类似于人类中栓塞疗法的肝动脉结扎术相结合,抑制了肝癌异种移植物的生长。
因此,本发明涉及作为趋化因子受体拮抗剂的肽,任选与一种或多种其他化疗剂和/或放射治疗组合,在制备药物中的应用,所述药物用于在对象的栓塞疗法期间治疗癌症或者降低或停止肿瘤生长,其中所述肽与人类CXCR4特异性结合。
本发明还涉及在对象中治疗癌症或者降低或停止肿瘤生长的方法,所述方法包括在栓塞疗法期间向所述对象施用任选与一种或多种其他化疗剂和/或放射疗法组合的肽,其中所述肽与人类CXCR4特异性结合。
优选地,所述肽还包含:
(1)由SEQ ID NO.1、2或3表示的氨基酸序列,或
(2)通过在SEQ ID NO.1、2或3中的一个或几个保守性替换得到的氨基酸序列。
优选地,所述肽可以由SEQ ID NO.1、2或3的氨基酸序列、或其由一个或几个保守性替换(例如1、2、3、4、5、6、7、或8个保守性替换)产生的变体的氨基酸序列组成。
优选地,所述对象是人类。
在一种实施方式中,所述化疗剂是选自抗有丝分裂剂、铂基化疗剂、受体酪氨酸激酶抑制剂、嘧啶类似物、拓扑异构酶抑制剂和佐剂中的至少一种。
优选地,所述抗有丝分裂剂是多西紫杉醇或紫杉醇,或其可药用的类似物或盐;所述铂基化疗剂是顺铂、卡铂、异丙铂或奥沙利铂,或其可药用盐;所述受体酪氨酸激酶抑制剂是索拉非尼、舒尼替尼或帕唑帕尼,或其可药用盐;所述嘧啶类似物是吉西他滨、5-FU或卡培他滨,或其可药用盐;所述拓扑异构酶抑制剂是依立替康、托泊替康、喜树碱或片螺素D,或其可药用盐;和所述佐剂是亚叶酸,或其可药用盐。
或者,所述化疗剂是5-FU、亚叶酸和奥沙利铂的组合;5-FU、亚叶酸和依立替康的组合;卡培他滨和奥沙利铂的组合;或顺铂和吉西他滨的组合。
在一种实施方式中,放射线是X射线、γ射线或带电粒子,其由身体外的机器或由置于身体内癌症/肿瘤细胞附近的放射性材料递送。优选地,所述放射性材料是放射性碘。
在一种实施方式中,所述癌症选自卵巢癌、子宫癌、乳腺癌、肺癌、肝癌、结肠直肠癌、膀胱癌、肾癌、前列腺癌、胰腺癌、胃癌、骨癌、皮肤癌、内脏癌、肾上腺和嗜铬细胞瘤、白血病和恶性软组织肉瘤。
在一种实施方式中,所述肽和化疗剂同时施用。在另一种实施方式中,所述肽和化疗剂相继施用。
在一种实施方式中,所述栓塞疗法是经动脉栓塞(TAE)、经动脉化疗栓塞(TACE)或放射性栓塞(RE)。
具体而言,本发明提供了CXCR4拮抗剂/抑制剂在栓塞疗法中的新治疗性应用,即本发明提供了联合疗法。在某些实施方式中,CXCR4拮抗剂可以如下治疗性使用,或者用于制造用于这样的治疗处理的药物:治疗癌症和调节血管生成。在本发明的一些方面,在栓塞疗法程序期间,可以在用或者不用另一种化疗剂的情况下使用CXCR4抑制剂。本发明提供了相应的医疗方法,其中以可药用的制剂形式施用治疗剂量的CXCR4拮抗剂。因此,本发明还提供了药物组合物,其包含CXCR4拮抗剂和本领域中常规的或如下所述的可药用赋形剂或载体。所述药物组合物可以有利地溶解在生理可接受的pH下的水溶液中。
用于本发明中的CXCR4拮抗剂可以是包含SDF-1的基本纯化的肽片段、修饰片段、类似物或可药用盐的肽化合物。在一些实施方式中,所述肽化合物可以包含N端氨基酸序列:KGVSLSYRC-X(SEQ ID NO.3),其中X选自氢和与SDF-1的至少一部分同源的多肽。
在另一种实施方式中,所述肽化合物可以包含二聚化的N端氨基酸序列(这里用从羧基端到氨基端书写的第二个二聚体表示):KGVSLSYR-X-RYSLSVGK(SEQ ID NO.1,名为CTCE-9908),其中X可以是赖氨酸,其中α-和ε-氨基二者都与同精氨酸残基形成酰胺键有关并且赖氨酰羧基可以通过乙酸反应(acetate acid reaction)被保护。在又一种实施方式中,所述肽化合物还可以包含二聚化的N端氨基酸(这里用从羧基端到氨基端书写的第二个二聚体表示):KGVSLSYRC-X-CRSLSVGK(SEQ ID NO.2),其中X可以是赖氨酸,其中α-和ε-氨基二者都与同半胱氨酸残基形成酰胺键有关并且赖氨酰羧基可以通过乙酸反应被保护。或者,在前述的二聚化肽化合物中,X可以是任何桥形成部分,其共价连接肽,使得多个肽通过所述桥相连以在所述化合物中提供多个N端。
根据本发明的一个方面,CXCR4的拮抗剂可以被治疗性用于在人类病理性疾病包括癌症例如淋巴瘤和癌瘤以及再狭窄中调节血管生成和细胞生长。在一种实施方式中,如本文中例示的,所述肽CXCR4拮抗剂已经用于在哺乳动物癌症的小鼠模型中抑制血管生成和肿瘤生长。
在各个方面,本发明运用了CXCR4拮抗剂。在一些实施方式中,用于本发明中的CXCR4拮抗剂可以是SDF-1α或SDF-1β的基本纯化的肽片段、修饰的肽片段、类似物或可药用盐。CXCR4的SDF-1衍生肽拮抗剂可以通过已知的生理测定法和各种合成技术鉴定(例如Crump等,1997,The EMBO Journal 16(23)6996-7007;和Heveker等,1998,Current Biology 8(7):369-376中公开的;其各自通过引用结合在本文中)。SDF-1的这样的类似物包括天然SDF-1的同系物,例如天然存在的同工型或遗传性变体,或与SDF-1具有显著的序列相似性的多肽,例如与天然SDF-1序列的至少一部分具有40%序列同一性、60%序列同一性或优选80%序列同一性,前提是它们具有CXCR4拮抗活性。在一些实施方式中,化学上相似的氨基酸可以取代天然SDF-1序列中的氨基酸(以提供保守性氨基酸替换)。
预期通过添加、缺失或取代一个或几个氨基酸残基,SEQ ID NO.1、2或3可具有变体,而这对于它们作为趋化因子受体拮抗剂、尤其是作为CXCR4拮抗剂的活性没有任何不良影响。在SEQ ID NO.1、2或3中有一个、两个、三个、乃至四个、最多八个氨基酸残基的各种保守性替换。例如,所述保守性替换可以发生在酸性氨基酸(例如Glu和Asp)、碱性氨基酸(例如Lys和Arg)、羟基氨基酸(例如Ser和Thr)、或芳族氨基酸(例如Phe、Trp和Tyr)当中。特别是,SEQ ID NO.1或2的X可以是Lys或Arg。
在具体的实施方式中,CXCR4拮抗剂的治疗或预防有效量的优选范围可以是0.01nM-0.1M,特别是0.1nM-0.05M,更特别是5nM-15mM和最特别是0.1mM-10mM。要注意,剂量值可以随着要缓解的病情的严重性而变化,尤其对于多发性硬化。要进一步理解,对于任何具体的对象,应该根据个人需要和管理或监督所述组合物给药的人员的专业判断,随时间调节具体的剂量方案,而且在本文中列出的剂量范围仅仅是示例性的,并不打算限制所述组合物的范围或实践。
所述组合物中活性化合物的量可以根据诸如个体的疾病状态、年龄、性别和体重这样的因素变化。可以调节剂量方案以提供最佳治疗响应。例如,可以施用单次大剂量,可以随时间施用几个分剂量,或者如治疗情况的紧急性所指示,所述剂量可以按比例降低或增加。尤其有利的是配制剂量单位形式的肠胃外组合物以便于剂量的施用和统一性。剂量单位形式用在本文中时是指适合作为用于待治疗哺乳动物对象的单位剂量的物理分立单位;每个单位含有与所需药物载体结合的经计算产生期望治疗效果的预定量活性化合物。本发明的剂量单位形式的规格由以下因素决定并直接取决于(a)活性化合物的独特特性和准备达到的具体治疗效果,和(b)本领域中混合这样的活性化合物来处理个体敏感性的固有的局限性。
用在本文中时,术语“可药用载体”或“赋形剂”包括生理相容的任何和所有的溶剂、分散介质、包衣、抗细菌和抗真菌剂、等渗和吸收延迟剂等。在一种实施方式中,所述载体适合于肠胃外给药。或者,所述载体可以适合于静脉内、腹膜内、肌内、舌下或口服给药。可药用载体包括无菌水溶液或分散体和用于临时制备无菌注射溶液或分散体的无菌粉末。将这样的介质和试剂用于药用活性物质是本领域中公知的。除非是在任何常规介质或试剂与活性化合物不相容的情况下,其在本发明的药物组合物中的使用就在考虑之内。补充的活性化合物(例如,一种或多种其他化疗剂)也可被纳入到所述组合物中。
本发明的肽可以利用标准技术化学合成,例如在Bodansky,M.肽合成原理(Principles of Peptide Synthesis),Springer Verlag,Berlin(1993)和Grant,G.A.(ed.).合成肽:使用者指南(Synthetic Peptides:A User's Guide),W.H.Freeman and Company,New York(1992)中描述的那些技术(它们全部通过引用结合在本文中)。自动肽合成仪是可商购的(例如,Advanced ChemTech Model 396;Milligen/Biosearch 9600)。
另外,本发明的肽可以根据标准重组DNA技术利用编码所述肽的核酸分子制备。编码所述肽的核苷酸序列可以利用遗传密码确定,并且具有这种核苷酸序列的寡核苷酸分子可以通过标准DNA合成方法(例如使用自动DNA合成仪)合成。或者,编码肽化合物的DNA分子可以根据标准分子生物学技术衍生自天然前体蛋白基因或cDNA(例如,利用聚合酶链反应(PCR)和/或限制性内切酶消化)。
附图说明
图l:SEQ ID NO.1肽二聚体(也称为CTCE-9908)与肝动脉结扎相结合(HAL+CTCE-9908),降低了肝细胞癌的肿瘤大小。以CTCE-9908与肝动脉结扎相结合(HAL+CTCE-9908)处理的大鼠显示肿瘤消退高达80%。这通过发光评估和尸检二者来看很明显。
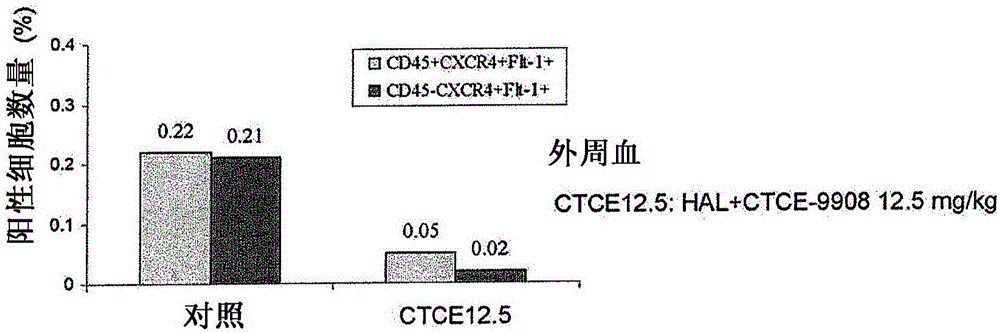
图2:CTCE-9908与肝动脉结扎结合,减少了循环血管细胞(hemangiocyte)的数量。与单独肝动脉结扎相比,用HAL和12.5mg/kg的CTCE-9908处理减少了CXCR4+Flt-1+血管细胞约80%至90%。减少这些细胞的募集可以促进肿瘤缩小。
图3:CTCE-9908在有或没有肝动脉结扎(HAL)下均显著增加大鼠肝细胞癌(HCC)模型的存活率。单独的HAL和假手术不显著影响HCC大鼠的存活率。单独的CTCE-9908以25mg/kg处理与单独的HAL和假手术相比,显著增加存活率。与单独的CTCE-9908相比,12.5mg/kg和25mg/kg的CTCE-9908二者与HAL相结合均进一步增加存活率。
具体实施方式
本发明将通过参考以下实施例进一步说明。要理解本发明不限于所述实施例。
实施例1
本实施例利用大鼠肝动脉结扎模型显示了CXCR4拮抗剂对肿瘤生长的抑制效应。
所使用的CXCR4拮抗剂是短肽二聚体拮抗剂CTCE-9908(SEQ ID NO.1)。大鼠原位肝细胞癌(HCC)模型通过将大鼠HCC细胞系CRL-1601注射到Buffalo大鼠肝脏左叶来建立。因为所述大鼠HCC细胞系是从Buffalo大鼠产生的,所以在Buffalo大鼠中体内形成的肿瘤可以模拟临床情况。肿瘤细胞注射后两周,将大鼠随机分成以下组:
1)假手术,n=6;
2)肝动脉结扎,n=6;
3)CTCE-9908,12.5mg/kg/天,n=6;
4)CTCE-9908,25mg/kg/天,n=6;
5)肝动脉结扎与CTCE-9908 12.5mg/kg/天相结合,n=6;
6)肝动脉结扎与CTCE-9908 25mg/kg/天相结合,n=6。
天然缺氧普遍出现在大的HCC肿瘤结节中。肝动脉结扎术是要模拟栓塞术治疗途径,栓塞术是不可切除的HCC最常用的治疗。这种治疗途径一方面导致肿瘤细胞坏死,另一方面它引起对残留肿瘤细胞的更严重的缺氧条件,导致促血管生成因子释放和随后肿瘤再生长。因此,在该提议的研究中,我们在有或没有肝动脉结扎下应用CTCE-9908,以探查这种药物单独使用或与缺血性缺氧相结合对于治疗HCC的效应。所述CRL-1601HCC细胞系是CXCR4阴性的。因此,CTCE-9908不会对肿瘤细胞本身具有直接效应。
第一组(对照)接受假手术继之以水灌注。第二、第五和第六组给予肝动脉结扎。第三和第五组每天皮下给予12.5mg/kg剂量的CTCE-9908。第四和第六组每天皮下给予25mg/kg剂量的CTCE-9908。持续处理4周。最后一次处理后一星期,对照和处理组二者均被处死。测量原发肿瘤的尺寸以及肝功能。通过免疫组织化学分析原发肿瘤和转移瘤中的微血管密度、肿瘤细胞增殖指数、肿瘤细胞的凋亡状态和形态。肺用福尔马林固定,石蜡包埋,连续切片和安装在载玻片上。分析转移性肿瘤灶的数量。通过ELISA分析血浆SDF-1。收集血液,通过流式细胞术分析血管细胞和内皮祖细胞。
本研究考查了模拟临床情况下经动脉化疗栓塞的大鼠肝细胞癌(HCC)模型。大鼠HCC植入肝脏左叶并使其生长2周,此时肿瘤块直径大于10mm。然后给予大鼠肝动脉结扎(HAL),并以12.5mg/kg/天或25mg/kg/天的CTCE-9908处理四周。最后一次CTCE-9908处理后一周,处死大鼠并分别通过发光和流式细胞术进行肿瘤以及循环细胞的评估。
所有大鼠对CTCE-9908处理耐受良好。CTCE-9908伴HAL导致肿瘤消退高达80%(图1)。观察到循环血管细胞减少(约80%-90%)(图2)。这可以通过抑制血管发生和血管生成来显著促进肿瘤缩小。大鼠存活率相应地增加。与假手术相比,单独的HAL不改变存活率。单独的CTCE-9908显著增加存活率。CTCE-9908与HAL结合进一步提高存活率(图3)。
实施例2
在遵循每个群组每个位置1个对象的加速滴定设计并遵循100%剂量增量的开放性剂量探索试验期间,不仅证明了用CXCR4受体拮抗剂化合物治疗各种实体瘤的安全性,而且证明了一定疗效。根据所述研究设计,所述群组将在中等毒性的情况下扩大,以包括更多的对象和额外的剂量增量(40%),直到达到5mg/kg的最大剂量或剂量限制性毒性。在剂量递增期间,仅在5mg/kg群组中观察到1例中等毒性,因此在标准期中的患者全部用5mg/kg治疗。
对象每天给药,连续4周(不包括周末和假期),并在对象完成他们的治疗后评价剂量限制性毒性。退出研究并且没有接受至少16剂研究药物的对象以相同的剂量水平替换,以便确保在加速期中每个群组至少一个对象完成治疗。
在所述研究期间报道的毒性根据国家癌症学会(National Cancer Institute)(NCI)CTCAE(不良事件常用术语标准)v3.0分级。剂量限制性毒性(DLT)如下定义:
在4周治疗期间,a)任何等级≥3可逆的非血液治疗相关的毒性,或
b)任何不可逆等级≥2的非血液治疗相关的毒性,或
c)任何等级≥4、持续长于10天的血液治疗相关毒性。
就本研究的目的而言,中等毒性根据NCI CTCAE v3.0定义。当发生不良事件时对它们进行处理。在对象处于所述研究中的时期期间,不施用意在治疗对象的恶性肿瘤的试验性或商业性药剂或疗法。
在研究期间观察到总共八例中等毒性,同时没有报道DLT。因此,在本研究期间,在患有难治性肿瘤的对象中,没有确立最大耐受剂量(MTD)。进一步的剂量递增可以确定MTD。正如许多靶向疗法一样,MTD可能永远达不到,并且可能不是生物有效剂量。然而,在本研究中观察到许多疗效趋势:五个对象在他们最后的治疗周期结束时总体病情稳定,包括在一个小肠对象中长达七个月的病情稳定,并且在一个卵巢癌对象和一个结肠直肠癌对象中肿瘤标记物降低。这些在1、2.5和5mg/kg/天剂量下观察到。这些剂量可以考虑用于2期研究。将来的研究可以考虑CTCE-9908与其他疗法相结合使用或用于其他患者群体。
参考文献
- 还没有人留言评论。精彩留言会获得点赞!