作为ALK/FAK/IGF1R多激酶抑制剂的吡咯并嘧啶衍生物的药物制剂组合物及其制备方法与流程
本发明涉及一种吡咯并嘧啶衍生物的药物制剂。具体地,本发明涉及alk/fak/igf1r多激酶抑制剂n-异丙基-2-((2-((2-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)-6,7-二氢-5h-吡咯并[2,3-d]嘧啶-4-基)氨基)苯磺酰胺或其药学上可接受的盐用于临床的稳定制剂,如片剂、胶囊剂或颗粒剂。
背景技术:
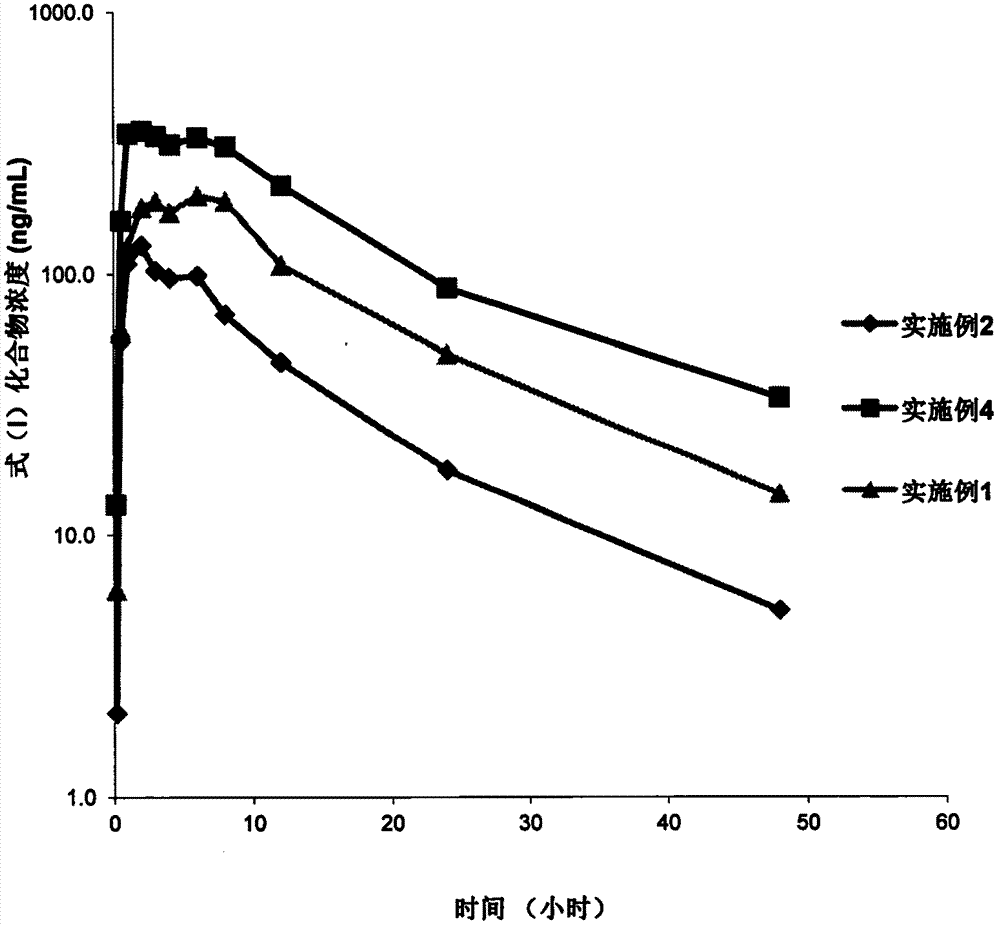
:式(i)化合物n-异丙基-2-((2-((2-甲氧基-4-(4-(4-甲基哌嗪-1-基)哌啶-1-基)苯基)氨基)-6,7-二氢-5h-吡咯并[2,3-d]嘧啶-4-基)氨基)苯磺酰胺是一种alk、fak、igf1r多激酶抑制剂,这些靶标与肿瘤的发生、发展均有密切关系。申请号为201110002776.3和201280004821.1的专利均已描述了此化合物在治疗哺乳动物(优选人)的肿瘤生长和癌症转移方面应用的良好前景。适用于制备式(i)化合物的口服固体制剂受限于式(i)化合物游离碱的差水溶性、弱碱性以及易氧化性。例如在胃的环境中(ph约1-2),式(i)化合物具有约30mg/ml的饱和溶解度,随着ph的升高,式(i)化合物的溶解度呈指数急剧下降,至ph6.8时溶解度仅为约4μg/ml,这意味着溶解于胃液中的式(i)化合物转移至肠道中时,显著量的式(i)化合物可能会析出,吸收减少,进而降低其生物利用度。另外,化合物结构中含有大量的仲胺和叔胺,导致其在生产和贮存中,极易氧化,产生降解杂质。较低的生物利用度和易氧化的不稳定性是式(i)化合物制剂面临的挑战。本发明旨在提供临床上使用安全、有效、质量可控的式(i)化合物制剂以及制备方法。提高其体内生物利用度,并且将主要由氧化产生的降解杂质控制在安全范围内。具体涉及片剂、胶囊剂或颗粒剂,尤其是片剂处方及其制备方法,药物和羟丙甲纤维素和或羟丙甲纤维素-衍生物聚合物的新型组合物以及制备方法。技术实现要素:本发明提供一种式(i)化合物制剂组合物以及制备方法。所述制剂组合物包含式(i)化合物或其可药学上可接受的盐,还包含填充剂、崩解剂、表面活性剂和润滑剂中的至少一种。填充剂又称为稀释剂,包括对于药学领域人员显而易见的淀粉、蔗糖、糊精、预胶化淀粉、微晶纤维素、无机盐类以及糖醇类等。在本发明的一个优选实施例中所选填充剂是微晶纤维素ph102。由于式(i)化合物结构中含有仲胺基团,为了避免可能发生的美拉德反应,处方中应该尽量避免使用乳糖。崩解剂包括干淀粉、羧甲淀粉钠、低取代羟丙基纤维素、交联羧甲基纤维素钠和交联聚维酮。本发明的一些实施例中优选崩解剂是交联羧甲基纤维素钠。表面活性剂包括离子型表面活性剂和非离子型表面活性剂。离子型表面活性剂主要包括阴离子表面活性剂,如高级脂肪醇硫酸酯、脂肪族磺酸化物、烷基芳基磺酸化物和烷基萘磺酸化物,非离子型表面活性剂包括环氧乙烷和环氧丙烷的嵌段共聚物、饱和或者不饱和的c8-c20酸的乙二醇或甘油酯、饱和或不饱和的c8-c20酸的聚氧乙烯酯、和/或不饱和的c8-c20酸的聚氧乙烯醚、饱和或不饱和的c10-c20酸的聚乙烯醇或脱水山梨醇酯。本发明的一些实施例中表面活性剂优选十二烷基硫酸钠,一些实施例中表面活性剂优选泊洛沙姆。在一些实施例中,表面活性剂可显著提高式(i)化合物片的体外溶出度和体内生物利用度。润滑剂包括对于药学领域人员显而易见的硬脂酸镁、硬脂酸、微粉硅胶、滑石粉、氢化植物油、聚乙二醇类、月桂醇硫酸钠、月桂醇硫酸镁、硬脂酸富马酸钠。式(i)化合物片的制备工艺包括对于药学领域人员显而易见的干法制粒、湿法制粒或者直接压片工艺。本发明还提供了一种式(i)化合物和羟丙甲纤维素和/或羟丙甲纤维素-衍生物的组合物及其制备方法。优选以下至少一种规格的羟丙甲纤维素:(i)hpmchme15cp;(ii)hpmchme100cp;(iii)hpmchme4m。优选羟丙甲纤维素-衍生物为醋酸羟丙甲纤维素琥珀酸酯(hpmcas)。在使用hpmcas的实施例中,优选下列至少一种作为hpmcas聚合物:(i)平均7重量%乙酰基含量和16重量%琥珀酰基含量;(ii)平均9重量%乙酰基含量和12重量%琥珀酰基含量;(iii)平均12重量%乙酰基含量和6重量%琥珀酰基含量。hpmcas的玻璃化转变温度(tg)约120℃。实施例中优选包括使用hpmcas:式(i)化合物的重量比由95%∶5%,至约50%∶50%。实施例中优选使用hpmcas:式(i)化合物的重量比为约66.7%∶33.3%。在实施例中,制备此组合物的方法包括:(i)混合式(i)化合物和hpmcas形成混合物;(ii)将混合物加热至一定温度(约60~170℃),形成溶解于hpmcas中的熔融分散体;(iii)冷却熔融分散体;(iv)经由口膜,将分散体挤成条状物;(v)条状物经由传送带传送至拉条造粒机,切碎,得到合适粒度的丸粒;(vi)丸粒和对于药物制剂领域人员显而易见的填充剂、崩解剂和润滑剂等辅料制备成片剂、胶囊剂或者颗粒剂。值得注意的是,由于式(i)化合物化学结构中含有大量的仲胺和叔胺,导致其在生产和贮存中,受氧气和光线的影响,极易氧化,产生降解杂质。在处方中尽可能避免使用乳糖,以避免可能发生的美拉德反应产生降解杂质。同时式(i)化合物片进行薄膜包衣,以避光和防潮,提高其化学稳定性。本发明还描述了一种式(i)化合物胶囊剂和颗粒剂以及制备方法。式(i)化合物装在胶囊壳中与外界隔离,减缓了空气、光线的影响,有利于降低稳定性期间式(i)化合物可能发生的降解反应,对式(i)化合物在一定程度上起了保护和稳定的作用。同时胶囊中的药物是以颗粒状态直接填充于囊壳中,在胃肠道中迅速分散、溶出和吸收。相同的,式(i)化合物的聚酯/铝/聚乙烯复合膜袋,hdpe瓶或双铝包装形式也使得其与外界的水分、空气和光线隔绝,稳定和保护药物,减少降解杂质,同时颗粒剂也具有分散和溶出迅速的优点。附图说明图1为在0.1n盐酸介质中的溶出曲线图。实施例1在0.1n盐酸介质中溶出迅速,10分钟溶出度即达85%以上。图2为在ph6.8磷酸盐缓冲液中的溶出曲线图。实施例1在ph6.8磷酸盐缓冲液中溶出极为缓慢,120分钟溶出度不超过4%。图3为实施例1、实施例2在ph4.5+0.3%sls中的溶出曲线图。与实施例2相比,实施例1在ph4.5磷酸盐缓冲液+0.3%sls介质中10分钟溶出度快约20%,60分钟溶出度快约10%。实施例1处方中含有表面活性剂十二烷基硫酸钠,而实施例2中不含表面活性剂,说明表面活性剂有助于改善式(i)化合物的疏水性,提高体外溶出度。图4为实施例4在0.1n盐酸中的溶出曲线图。实施例4在0.1n盐酸介质中120分钟溶出小于10%。这是因为hpmcas为肠溶型高分子聚合物,其在ph5.5以下不溶解,而在ph5.5以上溶解。图5为实施例4在ph6.8磷酸盐缓冲液中的溶出曲线图。实施例4在ph6.8磷酸盐缓冲液介质中40分钟达到最高累积溶出度约90%,120分钟溶出度约70%。说明hpmcas显著提高了式(i)化合物的溶解度,使其处于过饱和状态。图6为实施例1加速试验在ph4.5+0.3%sls中的溶出曲线图。实施例1经加速条件(40±2℃,75±5%rh)放置3个月,在ph4.5磷酸盐缓冲液+0.3%sls介质中的溶出与0月、1月相比无变化,溶出度稳定。图7为实施例1、实施例2和实施例4进行比格犬体内pk结果图。将实施例1、实施例2和实施例4进行比格犬体内pk研究。实施例1的auc0-48h是实施例2的2.22倍,说明表面活性剂能够显著提高体内生物利用度。实施例4的auc0-48h是实施例1的1.87倍,说明采用hpmcas制备的组合物制剂能够显著提高生物利用度。具体实施方式以下通过实施例进一步说明本发明,但不作为对本发明的限制。实施例1式(i)化合物片处方如下:原辅料名称用量(%,w/w)式(i)化合物10二氧化硅5微晶纤维素ph10275交联羧甲基纤维素钠3十二烷基硫酸钠6硬脂酸镁1包衣粉3式(i)化合物片制备工艺如下:1)将80g式(i)化合物和40g二氧化硅混合后过40目筛。其中式(i)化合物的粒度为d50约7.892μm,d90约19.172μm。2)将已过筛的式(i)化合物与二氧化硅的混合物与600g微晶纤维素ph102、24g交联羧甲基纤维素钠、48g十二烷基硫酸钠、4g硬脂酸镁置于hsd30料斗混合机(浙江迦南制药设备有限公司)中,设置转速为12rpm,混合10分钟。3)将物料加入gl2-25型干法制粒机(张家港市开创机械制造有限公司)料斗中进行干法制粒,设置送料频率为6~30hz,压片频率为10~30hz,制粒频率为10~20hz。4)将颗粒和4g硬脂酸镁置于料斗混合机中,设置转速为12rpm,混合5分钟。5)将物料采用旋转压片机压片。6)用欧巴代ii包衣,包衣增重为2~3%。7)实施例2式(i)化合物片处方如下:原辅料名称用量(%,w/w)式(i)化合物28.5二氧化硅5微晶纤维素ph10260.5交联羧甲基纤维素钠5硬脂酸镁1包衣粉3式(i)化合物片制备工艺如下:1)将228g式(i)化合物和40g二氧化硅混合后过40目筛。其中式(i)化合物的粒度与实施例1相同。2)将已过筛的式(i)化合物与二氧化硅的混合物与484g微晶纤维素ph102、40g交联羧甲基纤维素钠、4g硬脂酸镁置于hsd30料斗混合机中,设置转速为12rpm,混合10分钟。3)将物料加入gl2-25型干法制粒机料斗中进行干法制粒,设置送料频率为6~30hz,压片频率为10~30hz,制粒频率为10~20hz。4)将颗粒和4g硬脂酸镁置于hsd30料斗混合机中,设置转速为12rpm,混合5分钟。5)将物料采用旋转压片机压片。6)用欧巴代ii包衣,包衣增重为2~3%。实施例3式(i)化合物片处方如下:式(i)化合物片制备工艺如下:1)称取24g聚维酮k30和24g泊洛沙姆188,溶于适量纯化水中配成粘合剂溶液;2)将222.4g式(i)化合物、24g二氧化硅、297.6g微晶纤维素ph101以及24g交联羧甲基纤维素钠置于g6型湿法混合制粒机(深圳信宜特科技有限公司)中,设置搅拌桨转速2r/sec,剪切速度15r/sec,混合10分钟;3)喷入粘合剂溶液进行湿法制粒,雾化压力约0.4mpa,经20目筛制粒;4)60℃干燥湿颗粒至水分小于2%,用30目筛整粒;5)将颗粒和144g微晶纤维素ph102、32g交联羧甲基纤维素钠置于hsd30料斗混合机中,设置转速为12rpm,混合5分钟。再加入8g硬脂酸镁,混合5分钟。6)将物料采用旋转压片机压片。7)用欧巴代ii包衣,包衣增重为2~3%。实施例4式(i)化合物片处方如下:式(i)化合物片制备工艺如下:1)将200g式(i)化合物和400ghpmcas置于hsd30料斗混合机中混合,转速为12rpm,混合5分钟。其中式(i)化合物的粒度为d50约28.806μm,d90约86.543μm;2)将上述混合物加入process11型热熔挤出机(thermofisher)的喂料器中,加热至125~135℃,形成熔融分散体,冷却该熔融分散体,然后经由孔径为2mm的口模挤出条状物;3)条状物继续冷却后,经由锤式粉碎机锤磨,得到平均粒径为d50约70~400μm的颗粒;4)将颗粒和40g二氧化硅、112g微晶纤维素ph102、40g交联羧甲基纤维素钠置于料斗混合机中,设置转速为12rpm,混合5分钟。再加入8g硬脂酸镁,混合5分钟。5)将物料采用旋转压片机压片。6)用欧巴代ii包衣,包衣增重为2~3%。实施例5式(i)化合物颗粒处方如下:原辅料名称用量(%,w/w)式(i)化合物75预胶化淀粉10微晶纤维素ph10210交联羧甲基纤维素钠5式(i)化合物颗粒制备工艺如下:1)将750g式(i)化合物、100g预胶化淀粉、100g微晶纤维素ph102以及50g交联羧甲基纤维素钠置于hsd30料斗混合机中,转速为12rpm,混合10分钟。2)将物料加入gl2-25型干法制粒机料斗中进行干法制粒,设置送料频率为6~30hz,压片频率为10~30hz,制粒频率为10~20hz。3)将制得颗粒用聚酯/铝/聚乙烯复合膜袋进行内包装。实施例6式(i)化合物胶囊处方同实施例5,将实施例5中式(i)化合物颗粒200mg填充至2#胶囊壳中,得到规格为150mg的式(i)化合物胶囊。实验例将实施例1、实施例2和实施例4中的式(i)化合物片进行比格犬体内pk研究。采用交叉给药方式,将比格犬随机分成3组,每组6只,雌雄各半,体重在6.8~7.5kg,口服单次给药,给药剂量为150mg/只。3个实验组分别给实施例1、实施例2和实施例4中的式(i)化合物片。给药之后的0.17、0.5、1、2、3、4、6、8、12、24、48小时收集血样至肝素管中,血浆样品检测前在-80℃的温度下贮存。血浆中药物浓度采用hplc-ms/ms法测定。各个血药浓度-时间数据运用软件winnonlin模型进行药代动力学分析。拟合并计算主要药动学参数auc0-48h、cmax、tmax、和t1/2。药动学参数实施例1实施例2实施例4auc0-48h(hr*ng/ml)363816406813t1/2(hr)14.427.012.7cmax(ng/ml)229129379tmax(hr)2.543.043.00当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种快速检测BCR/ABL基因融合的探针组、试剂盒及方法与流程
- 酪氨酸激酶抑制剂的制造方法与工艺
- 芳香族酰胺类衍生物、其制备方法及其在医药上的应用与制造工艺
- 包含5α‑还原酶抑制剂的药物组合物的制造方法与工艺
- 作为脾酪氨酸激酶抑制剂的二氮杂螺烷酮取代的噁唑衍生物的制造方法与工艺
- 包含热弹性感觉剂和TRPA1受体抑制剂的皮肤接合剃刮助剂的制造方法与工艺
- 检测EGFR耐药性突变的方法和组合物与制造工艺
- 5‑溴‑7‑氮杂吲哚啉‑2‑酮类化合物及其制备方法与制造工艺
- 3‑[(3‑{[4‑(4‑吗啉基甲基)‑1H‑吡咯‑2‑基]亚甲基}‑2‑氧代‑2,3‑二氢‑1H‑吲哚‑5‑基)甲基]‑1,3‑噻唑烷‑2,4‑二酮和EGFR TYR激酶抑制剂的组合的制造方法与工艺
- 一种抗癌药物的晶体化合物及其制备方法与制造工艺