分子信标探针及引物对、VDR基因SNPs位点检测方法与流程
本发明属于基因检测技术领域,具体涉及一组分子信标探针及引物对、vdr基因snps位点检测方法。
背景技术:
维生素d受体(vitamindreceptor,vdr)是细胞表面结合1,25-二羟维生素d(1,25-dihyroxyvitamind,1,25(oh)2d,也叫钙三醇,calcitriol)的受体。当vdr与激素性质的1,25(oh)2d结合而活化后,便进入胞核内,与视黄醇-x受体形成异二聚体结合到特定基因的激素反应元件上,调节基因表达或转录抑制。因此,vdr也是一种核受体转录因子,即nr1i1(nuclearreceptorsubfamily1,groupi,member1),其调节的基因涉及钙磷代谢、细胞生长、增殖和凋亡,并进而与骨骼健康、免疫反应和肿瘤等过程相关。
vdr基因在人染色体上定位于12q13.11,具有500种以上的单核苷酸多态性位点和相应基因型,其中,apa1、cdx2、ecorv、bsm1、fok1、taq1、tru9i是早期采用限制性酶切片段长度多态性分析发现的snps,并且在过去一段时间积累了大量关于它们和多种健康与疾病现象相关的报道。vdr的rna产物由9个外显子构成,apa1位点(rs7975232)位于第8号内含子3’端,为[a/c]二态性,taq1位点(rs731236)位于第9号外显子5’端,为[c/t]二态性,二者相距较近。其中,在外显子上的taq1多态性并不改变对应的异亮氨酸,为同义突变。但位于内含子上的snps或位于外显子上氨基酸不变的snps可能与mrna转录、剪接、密码子翻译效率等过程相关,因此,仍可能影响疾病风险。
对apa1和taq1多态性的分析显示,二者与肺结核疗效、高加索人骨折与银屑病风险、胰岛素抵抗、哮喘和遗传过敏、腰椎病相关;taq1可能与男性帕金森病、汉族2型糖尿病合并胫前皮肤黑斑、骨密度、高体质指数、系统性红斑狼疮预后等疾病风险相关;apai与亚洲人的牙龈炎、我国北方和土耳其人的银屑病、肾细胞癌和散发性前列腺癌有关。因此,对人群的这两个snps进行检测,对分析vdr与多种疾病风险和表型的相关性具有研究与预测价值。
虽然等位基因检测的方法有数十种,但针对vdrapa1和taq1两个snps的基因型分析,目前报道的方法主要是:聚合酶链式反应后的限制性酶切片段长度多态性分析(pcr-rflp)、taqman探针、高分辨率熔解曲线分析(hrm)、多重pcr结合标签阵列单碱基技术、时间飞行质谱法、测序等。pcr-rflp虽然廉价,但其操作步骤繁琐,且需要两个独立的rflp实验进行基因分型,在分析大量样本时,尤其费时。以taqman探针不论混合还是单独反应分析这两个snps,都需精心设计、测试多套探针,增加了试剂成本。hrm技术存在部分非典型曲线难以判断的不足。时间飞行质谱、测序等方法都依赖于昂贵的设备、封闭的试剂和较高的实验操作技术,甚至需要摸索建立适宜的检测条件;且因其单次运行所需试剂用量有最低要求,间歇与随时开机运行的成本较高。
技术实现要素:
本发明的目的在于克服现有技术的上述不足,提供一组分子信标探针及引物对、vdr基因snps位点检测方法,旨在解决现有vdr基因snps位点检测步骤繁琐、耗时、成本高的技术问题。
为实现上述发明目的,本发明采用的技术方案如下:
一方面,本发明提供一组用于检测vdr基因snps位点的分子信标探针,包括第一探针和第二探针,所述第一探针用于检测vdr基因的多态性位点apa1,所述第二探针用于检测vdr基因的多态性位点taq1;其中,所述第一探针包含第一核心序列5’-tgggc[a/c]cctcactgctca-3’,所述第二探针包含第二核心序列5’-ggatggcctc[a/g]atcagc-3’;
同时,所述第一探针和所述第二探针的长度均为20-30bp,tm值均为62-65℃,所述第一探针的5’端和3’端分别用第一荧光分子和第一淬灭分子标记,所述第二探针的5’端和3’端分别用第二荧光分子和第二淬灭分子标记。
另一方面,本发明提供一组用于检测vdr基因snps位点的分子信标探针和引物对,所述分子信标探针为上述分子信标探针,所述引物对包括第一引物和第二引物,所述第一引物和所述第二引物的长度均为17-25bp;且所述第一引物包含5’-gttgagt-3’,所述第二引物包含5’-ggatgtac-3’。
再一方面,本发明提供一种用于检测vdr基因snps位点的试剂盒,包括上述分子信标探针和引物对,还包括taqhs酶、dntps、pcr缓冲液。
最后,本发明提供一种vdr基因snps位点的检测方法,包括如下步骤:
提取dna样本;
将所述dna样本与上述试剂盒配成pcr反应体系,进行pcr扩增;
根据所述扩增结果分析样本中vdr基因snps位点。
本发明提供的该组分子信标探针中,第一探针的核苷酸序列可覆盖vdr基因的多态性位点apa1(rs7975232),第二探针的核苷酸序列可覆盖vdr基因的多态性位点taq1(rs731236),这样,仅需要qpcr仪和该两条荧光标记的探针,即可实现对大量样本的vdr基因中该两个重要多态性位点的分析,加上qpcr仪运行成本和操作技术要求相对较低,这就大大降低了检测成本和分析难度。
同时,设计一组与该两条探针配套的引物对,一条(第一引物)位于apa1上游100bp范围内的特异性引物,另一条(第二引物)位于taq1下游100bp范围内的特异性引物。apa1上游100bp的适宜引物都有共同位置上7bp的片段5’-gttgagt-3’,taq1下游100bp的适宜引物都有共同位置上8bp的片段5’-ggatgtac-3’;这样可实现对大量样本的vdr基因中两个重要多态性位点的分析。
该用于检测vdr基因snps位点的试剂盒,因含有本发明特有的该组分子信标探针和引物对,所以具有高效、灵敏、特异性强的特点,而且该试剂盒成本低、使用方便,仅需要qpcr仪可实现对大量样本的vdr基因中两个重要多态性位点的分析。
该vdr基因snps位点的检测方法,因使用本发明特有的试剂盒,根据荧光信号在熔解曲线程序中出现的峰型判断vdr不同的基因型。因此具有检测步骤简单、耗时少、成本低的特点。
附图说明
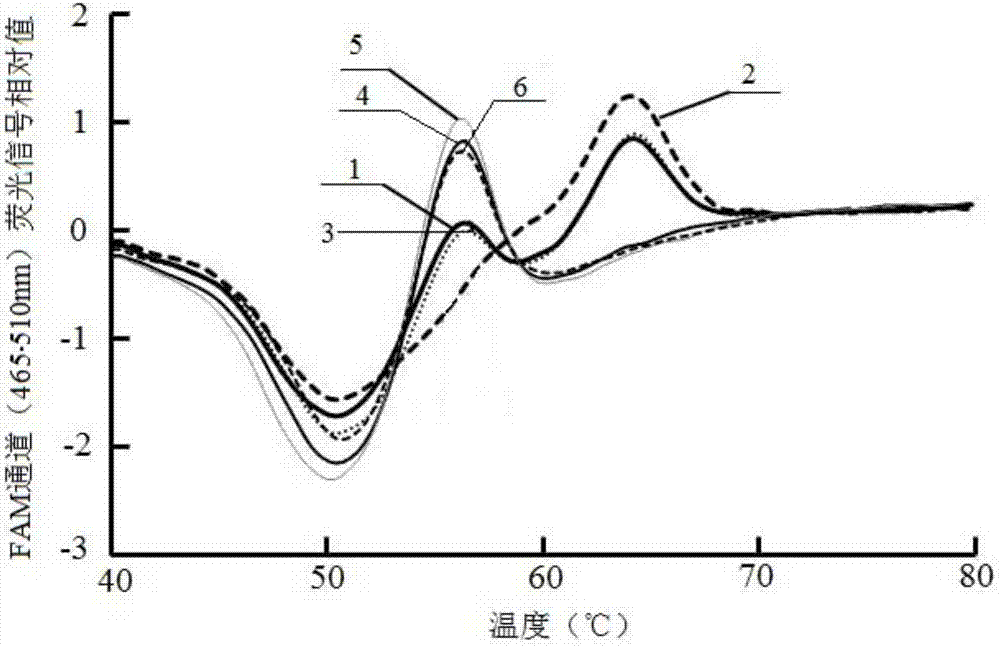
图1为本发明实施例2中fam通道荧光信号原始数据图;
图2为本发明实施例2中fam通道荧光信号原始数据处理图;
图3为本发明实施例2中hex通道荧光信号原始数据图;
图4为本发明实施例2中hex通道荧光信号原始数据处理图;
图5为本发明实施例2中pcr检测产物测序结果图;
其中,附图标记为:
1:一号样本(多态性:a/c-t/t);
2:二号样本(多态性:c/c-t/t);
3:三号样本(多态性:a/c-c/t);
4:四号样本(多态性:a/a-c/t);
5:五号样本(多态性:a/a-t/t);
6:六号样本(多态性:a/a-c/c)。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图和实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
一方面,本发明实施例提供了一组用于检测vdr基因snps位点的分子信标探针,包括第一探针和第二探针,所述第一探针用于检测vdr基因的多态性位点apa1,所述第二探针用于检测vdr基因的多态性位点taq1;其中,所述第一探针包含第一核心序列5’-tgggc[a/c]cctcactgctca-3’,所述第二探针包含第二核心序列5’-ggatggcctc[a/g]atcagc-3’;
同时,所述第一探针和所述第二探针的长度均为20-30bp,tm值均为62-65℃,所述第一探针的5’端和3’端分别用第一荧光分子和第一淬灭分子标记,所述第二探针的5’端和3’端分别用第二荧光分子和第二淬灭分子标记。
本发明是基于分子信标探针(molecularbeaconprobe)开发的一种同时检测apa1和taq1两个snps的熔解曲线分析技术。分子信标探针是一种呈发夹茎环结构的寡核苷酸探针,两个末端标记有荧光分子和淬灭分子,因探针两端的核酸序列互补配对而使这两个基团紧密相邻。荧光分子被激发后产生的光子被淬灭剂淬灭。定量聚合酶链式反应的熔解曲线程序中,茎环结构在加热变性后展开呈线性,并在退火阶段互补结合到目的序列上,使得荧光分子与淬灭分子分开,前者被激发出的荧光信号便可被qpcr仪记录。由于dna杂交双链碱基互补配对的密切程度(是否存在单碱基错配)会影响探针与目的序列杂交双链的熔解温度tm,因而snp的碱基情况便能被特定探针区分,即:探针与目的序列完全互补时,tm较高,随着错配碱基的增多,tm会降低;对于二态性(仅有2种碱基变化)的snp位点,纯合型二倍体为一个峰,杂合型二倍体呈现双峰。本发明实施例提供的该组分子信标探针中,第一探针的核苷酸序列可覆盖vdr基因的多态性位点apa1(rs7975232),第二探针的核苷酸序列可覆盖vdr基因的多态性位点taq1(rs731236),这样,仅需要qpcr仪和该两条荧光标记的探针,即可实现对大量样本的vdr基因中该两个重要多态性位点的分析,加上qpcr仪运行成本和操作技术要求相对较低,这就大大降低了检测成本和分析难度。
具体地,本发明实施例中,该两条探针的长度为20-30bp,而“环”一般长8-20个核苷酸,是与目标序列互补的探针主体,tm值比qpcr的退火温度高7-10℃,如推荐ta=55℃,探针的tm便在62-65℃;“茎”一般长5-7对核苷酸,可部分或全部由人为加入的碱基形成,cg碱基含量占75-100%,以使“茎”的tm比qpcr的ta高7-10℃,荧光分子一端不宜紧接g碱基,避免后者的荧光淬灭作用。在本发明一优选实施例中,第一探针的核苷酸序列为seqidno:1:cttgggcccctcactgctcaag;第二探针的核苷酸序列为seqidno:2:cgcggatggcctcaatcagcgcg。该优选的探针检测效果最佳。
具体地,在本发明实施例中,第一荧光分子和第二荧光分子均为fam、hex、tet、vic、rox、cy5、cy3、joe、alex和cal中的任意一种,且第一荧光分子和第二荧光分子为不同的荧光分子;而第一淬灭分子和第二淬灭分子均为dab、bhq、eclipse和tamra中的任意一种。在本发明一优选实施例中,第一荧光分子为fam,第二荧光分子为hex,第一淬灭分子和第二淬灭分子均为dab。
另一方面,本发明实施例还提供了一组用于检测vdr基因snps位点的分子信标探针和引物对,该分子信标探针为本实施例上述分子信标探针,而引物对包括第一引物和第二引物,该第一引物和第二引物的长度均为17-25bp;且第一引物包含5’-gttgagt-3’,第二引物包含5’-ggatgtac-3’。
具体地,在本发明实施例中,第二引物与第一探针和所述第二探针在同一链上,且第二引物的tm值为62-65℃,第一引物的tm值为58-60℃。一优选实施例中,第一引物为seqidno:3:gccgttgagtgtctgtgt;第二引物为seqidno:4:ggcggcagcggatgtacg。
又一方面,本发明实施例提供一种用于检测vdr基因snps位点的试剂盒,包括本发明实施例的分子信标探针和引物对,还包括taqhs酶、dntps、pcr缓冲液。该用于检测vdr基因snps位点的试剂盒,因含有本发明特有的分子信标探针和引物对,所以具有高效、灵敏、特异性强的特点,而且该试剂盒成本低、使用方便,仅需要qpcr仪可实现对大量样本的vdr基因中两个重要多态性位点的分析。
最后,本发明实施例提供一种vdr基因snps位点的检测方法,包括如下步骤:
s01:提取dna样本;
s02:将上述dna样本与本发明实施例的试剂盒配成pcr反应体系,进行pcr扩增;
s03:根据扩增结果分析样本中vdr基因snps位点。
该vdr基因snps位点的检测方法,因使用本发明实施例特有的试剂盒,根据荧光信号在熔解曲线程序中出现的峰型判断vdr不同的基因型,因此检测步骤简单、耗时少、成本低的特点。
具体地,上述pcr扩增为降落pcr扩增。
具体地,上述dna样本为人全血dna样本。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。本说明书中的英文缩写的全称和中文内容对应如下:
1)1,25(oh)2d:1,25二羟羟维生素d,1,25-dihydroxyvitamind
2)dab:二甲氨基偶氮苯甲酰(一种荧光淬灭剂),dabsyl
3)fam:羧基荧光素,carboxyfluorescein
4)hrm:高分辨率熔解曲线法,highresolutionmeltinganalysis
5)ncbi:美国国立生物技术信息中心,nationalcenterforbiotechnologyinformation
6)pcr:聚合酶链式反应,polymerasechainreaction
7)qpcr:定量pcr,quantitativepolymerasechainreaction
8)rflp:限制性酶切片段长度多态性,restrictionfragmentlengthpolymorphism
9)snp(s):单核苷酸多态性位点,singlenucleotidepolymorphism(s)
10)ta:退火温度,annealingtemperature
11)tm:熔解温度,meltingtemperature
12)vdr:维生素d受体,vitamindreceptor;斜体vdr表示其基因。
实施例1
1)获得目标snps基本信息
从文献获知vdrapa1的snp代码为rs7975232、vdrtaq1的snp代码为rs731236,从美国国立生物技术信息中心(nationalcenterforbiotechnologyinformation,ncbi)网站的snp数据库查询这两个snps位点信息如下:
vdrapa1,rs7975232[homosapiens]:
aaggcacaggagctctcagctgggc[a/c]cctcactgctcaatcccaccacccc;
vdrtaq1,rs731236[homosapiens]:
ctggggtgcaggacgccgcgctgat[c/t]gaggccatccaggaccgcctgtcca;
将上述两段序列输入nucleotideblast在线程序(database=humanrefseqgenesequence,optimizefor=或somewhatsimilarsequences(blastn)),与之匹配的片段位于ng_008731.1上的65003-64952(反向)和65032-65083,链接进入ng_008731.1的网页,获取64952-65083两侧适度(两个snps两侧各100bp)扩展的dna片段(如下所示),用于后续的引物和探针设计。从如下序列中可知,apa1和taq1二者间隔79bp,可采用一对引物扩增同时含这两个位点的dna片段,并在同一个反应体系中采用两种不同的荧光探针分析这两个二态性的snps。
2)探针与引物设计
本实施例中,分子信标探针和引物的设计总结为以下法则:
①“环”一般长8-20个核苷酸,是与目标序列互补的探针主体,tm值比qpcr的退火温度高7-10℃,如推荐ta=55℃,探针的tm便在62-65℃;
②探针上两端距离临近的snp应至少有3个碱基能与目的dna片段完全互补(即snp不应在靠近两端的3个碱基范围内),或至少2个c或g碱基与目的dan序列互补,以保证待测位点与探针不互补时,探针两端有一定的结合能力;
③“茎”一般长5-7对核苷酸,可部分或全部由人为加入的碱基形成,cg碱基含量占75-100%,以使“茎”的tm比qpcr的ta高7-10℃,荧光分子一端不宜紧接g碱基,避免后者的荧光淬灭作用;
④5’末端标记荧光分子(荧光报告基团),3’末端标记淬灭分子(荧光淬灭基团);
⑤整个分子信标探针全长一般20-30个核苷酸,不能与引物形成可以引发扩增的稳定二聚体,且探针和引物与目的dna的互补结合区域没有重叠部分;
⑥引物扩增目的dna片段的产物长度宜在200bp以内,最好短于150bp;
⑦对应于同一条dna单链上的引物和探针具有相近的tm值,且高于另一条引物的tm5-8℃;
⑧引物符合上述要求外,再符合其余的引物设计常规法则。
其中,⑤⑥⑦三条法则连同后文qpcr反应体系(见后文表5)中提高探针及其互补链引物浓度(即:提高探针互补链产量的非对称pcr)的策略,都有助于增强探针与目标链结合的竞争优势。类似地,也可根据正义链的互补dna链为模板设计探针。
以在线软件ncbiprimer-blast和primer3在apa1上游100bp范围内搜索上游引物、在taq1下游100bp范围内搜索下游引物,产物片段长200bp以内,对引物的位置做初步检索。以vdr的正义dna链为模板,代表性候选引物如表1所示。
表1
1粗体下划线或灰色涂抹字母代表同一引物群中,至少两个以上的引物的共有位置上的片段。上游引物群ii的共同片段也存在于表2;下游引物群ii中的灰色涂抹部分是与表2中下游引物的共同位置片段。
根据上述信息,再以primerpremier5软件在第一个snp上游3bp以外的97bp范围内设计上游引物,在第二个snp下游3bp以外的97bp范围内设计下游引物,引物长度预设(21±4)bp,产物长度预设100-200bp,搜索严格程度从“非常高(veryhigh)”开始,其余参数为默认值,直到“中等(morderate)”才出现5对引物,这些引物对的信息如表2所示。
表2
1以apa1上游100bp的碱基为1开始计算位置,后同。
2以tmutilityv1.3软件计算的tm值,条件为:引物0.2μm、目的dna0.2μm、mg2+2.0mm、总dntp0.8mm。
从表1和表2可见,apa1上游和taq1下游各100bp范围内,均有可能出现适宜的引物对。结合表1、表2的信息和引物、探针设计的要求,考虑将表2中的f2和r2组成引物对,在primerpremier5中手动调整和评估,得到的引物分别命名为vdr-f1和vdr-r1,见表3。
3)最佳探针与引物的理论筛选
由于下游引物tm为64.65℃,高于上游引物。根据上述探针与引物设计法则⑦,则探针与下游引物位于同一条链,即在负义链上设计探针,且探针与下游引物的tm接近。以primerpremier5软件手动选择分别覆盖apa1位点(对应64952-65083序列中的第101位碱基)和taq1位点(对应64952-65083序列中的第181位碱基)的探针。以primerexpress3.0的引物探针测试工具(primerprobetesttool)对引物对和探针进行分析,从自身发夹(hairpin)、自身二聚体(selfdimers)和交叉二聚体(crossdimers)的形成情况滤除欠佳的组合,针对待检的两个位点,可获得多条供选择的探针。得到引物序列和有待二级结构分析的探针见表3。
表3
1下划线或灰色涂抹的碱基是与表1、表2中上游或下游引物具有相同位置的片段,中括号内碱基为snps。
2以tmutilityv1.3软件计算的tm值,条件为:oligo0.2μm、目的dna0.2μm、mg2+2.0mm、总dntp0.8mm;探针的tm计算不含两端手动加入的碱基(小写字母)。
3探针3’端连接dab荧光淬灭分子;apa1-p的5’端连接fam荧光分子,taq1-p的5’端连接hex荧光分子。探针与下游引物对应于同一条dna链。
对分别覆盖apa1和taq1的探针,在其末端加入可以形成发夹结构的适当碱基,利用在线分析工具dnafoldingform进行“茎环”结构的质量评估。在特定熔解曲线分析条件下,分子构象合理而稳定(自由能差值dg<0)的分子信标探针,其预测的二级结构主要满足以下条件:①有正确的“茎”结构,保证荧光分子和淬灭分子空间靠近;②不宜出现7对以上碱基互补的“茎”结构。如不满足上述条件,需调整探针位置或/和构成“茎”的碱基。由于一条荧光探针的价格大约在900-1200元,因此,尽可能用软件和多方面信息手段对其事先做比较、评价,挑选最优的一条合成。本发明拟用熔解曲线分析的ta为40℃、na+50mm、mg2+2mm,该条件下,用dnafoldingform在线软件分析结果,表3中分子信标探针的合理而稳定的二级结构分析如下:其中小写字母为手动加入的碱基。
可见,表3中的探针序列符合设计要求,以该设计的探针要求探针合成公司在apa1-p的5’端连接fam荧光分子、taq1-p的5’端连接hex荧光分子,二者3’端均连接dab荧光淬灭分子。经过上述筛选得到的引物和探针对应于qpcr扩增片段(185bp)的位置如下:qpcr扩增产物全长185bp,灰色涂抹的碱基为上游引物序列和下游引物反向互补序列,方框内的碱基为探针对应位置(互补),中括号内的碱基为多态性位点。
此外,表1-表3中的全部引物(或其互补序列)和探针(或其互补序列)也可通过适当组合、评估,人工增减碱基,以达到本发明的目的,这里不再一一分析、验证,具体地,凡是具有表3中相同性能的引物和探针都在本发明保护范围内。
实施例2
1)dna样本制备:根据全血dna提取试剂盒(如北京天根生物技术有限公司的全血dna提取试剂盒,货号dp318-03)的操作说明提取待检测的dna样本,用微量分光光度计(如ge公司的nanovueplus)检测样本在280nm和260nm波长下的比值和由此吸光度值计算的dna浓度。
2)qpcr实验测试引物和探针:按表4中的qpcr成分配制反应试剂,并按表5中的qpcr反应程序进行预实验。三个推荐策略是:①热启动dna聚合酶,确保引物、探针分别与互补链杂交的特异性;②非对称产物扩增,提高一侧引物浓度以提高分子信标探针互补链的产量。③降落pcr(touch-down)程序提高扩增的特异性。
本实验的qpcr仪为roche480ii,用该仪器同时检测apa1和taq1组成的6种基因型(具体见表6的六个样本,实际情况不限于在此举例的6种组合),检测apa1位点的第一探针荧光信号检测波长为465-510nm(fam通道),检测taq1位点的第二探针荧光信号检测波长为533-580nm(hex通道),检测结果如图1-图4所示。图1为本实施例中fam通道荧光信号原始数据图;图2为本实施例中fam通道荧光信号原始数据处理图;图3为本实施例中hex通道荧光信号原始数据图;图4为本实施例中hex通道荧光信号原始数据处理图,图中6个标号分别对应六个不同样本检测到的荧光信号曲线。
从图1-图4可知,本实施例的vdr基因snps位点检测方法,只需要根据荧光信号在熔解曲线程序中出现的峰型,即可判断vdr的基因型是哪一种。对图1和图2检测的apa1位点,一、三号样本为a/c型,二号样本为c/c型,四、五、六号样本为a/a型;对图3和图4检测的taq1位点,一、二、五号样本为t/t型,三、四号样本为c/t型,六号样本为c/c型,结果见表6。因此,整个检测过程步骤简单、耗时少、成本低,在临床与预防医学的分子流行病学相关工作中非常实用。
表4
1taq热启动酶(takarataqtmhotstartversion)试剂盒,货号r007a,takara(大连)公司,含:takarataqhs酶、10×pcrbuffer(mg2+plus)和dntpmixture(各2.5mm)。
2分子信标探针与下游引物对应于同一条dna链上,分子信标探针浓度和上游引物的用量高于下游引物,确保在与上游引物pcr产物互补结合过程中,探针比下游引物pcr产物更具竞争优势,从而使探针互补的dna链有较高的产量,并能在qpcr仪的检测结果中呈现出与各种基因型一一对应的荧光信号峰型。
表5
1表中各中文名词对应于roche480iiqpcr操作软件的英文依次如下:程序名称(programname);分析模式(analysismode);循环数(cycles);目标温度(target);时间(hold);斜率(ramprate);第二目标温度(sectarget);步长(stepsize);信号采集次数(acquisitions);采集模式(acquisitionmode);预变性(pre-incubation);无(none);降落pcr(touch-down);扩增(amplification);定量(quantification);单次(single);熔解曲线(meltingcurve);连续(continuous);冷却(cooling)。
2仪器设置:检测模式(detectionformat)为多色探针(multicolorhydro-probe);定制(customize):荧光(fluos,465-510和533-580);模块规格(blocksize):96孔;反应体积(reactionvolume):25μl。
3表格中的“—”表示无需设置。
4连续监测40-80℃升温区间的荧光信号,分子信标探针从发夹结构打开,并结合到互补dna链上,荧光信号达到最大值。随着温度升高,有错配的探针-dna杂交链会在较低温度下解链,形成熔解曲线峰值;而匹配程度高的探针-dna杂交链会在较高温度下解链。
表6
3)将上述六个样本pcr产物送往商业服务公司进行测序,以验证分子信标qpcr的分型结果。6种基因型的样本各3个测序的结果与本发明技术分析的结果一致。图5将6种基因型的结果各列举了一例:图中
在研究vdr基因与人群多种慢性病的关系时,分析其apa1(rs7975232)和taq1(rs731236)的基因型是一项重要内容。利用本发明实施例的方法,我们曾分析了深圳市某人群3369名志愿者的这两个snps进行了分析,这两个snps基因型分布的hardy-weinberg平衡检验证实样本具有良好代表性。表7列举了现有文献中报道的我国人群和印度人、英国人中vdrapa1、taq1两个snps的基因型频率,以及我们应用本实施例检测到的基因型频率。结果显示:本发明实施例检测到的结果与此前我国人群对照组的研究数据一致(p>0.05),并显著不同于其他人种的情况(p<0.05),说明本技术在应用上有效。
表7
1apa1与同一文献中的对照组比较,p<0.05。
2taq1与同一文献中的对照组比较,p<0.05。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
sequencelisting
<110>深圳市慢性病防治中心
<120>分子信标探针及引物对、vdr基因snps位点检测方法
<160>4
<170>patentinversion3.3
<210>1
<211>22
<212>dna
<213>人工合成
<400>1
cttgggcccctcactgctcaag22
<210>2
<211>23
<212>dna
<213>人工合成
<400>2
cgcggatggcctcaatcagcgcg23
<210>3
<211>18
<212>dna
<213>人工合成
<400>3
gccgttgagtgtctgtgt18
<210>4
<211>18
<212>dna
<213>人工合成
<400>4
ggcggcagcggatgtacg18
- 还没有人留言评论。精彩留言会获得点赞!
- 一种快速区分prv中国型、欧美型及疫苗株的双色荧光检测方法、引物及探针的制作方法
- 实时荧光pcr技术定量检测myd88基因l265p突变的方法、引物和探针的制作方法
- 用于定性检测白血病融合基因的引物、探针、试剂盒及方法
- 在体外检测或/及定量乙型肝炎病毒的方法及其所使用的引物、探针的制作方法
- 用于检测多种呼吸道病毒的pcr引物组、探针组及试剂盒的制作方法
- 一种检测人类met基因14外显子剪接突变的引物、探针及试剂盒的制作方法
- 使用重叠引物和熔解探针检测单核苷酸多态性的制作方法
- 用于检测多种呼吸道病毒的pcr引物组、探针组及试剂盒的制作方法
- 一种检测人类met基因14外显子剪接突变的引物、探针及试剂盒的制作方法
- 使用重叠引物和熔解探针检测单核苷酸多态性的制作方法