作为生物标记物的小ncRNA的制作方法
本发明提供了作为生物标记物的小ncrna,其用于对个体的健康状况进行分类。本发明还提供了鉴定ncrna生物标记物的筛选方法。
背景技术:
人类基因组编码大量的小非编码蛋白质的rna(ncrna)转录物。已经描述了多种ncrna类型,包括高丰度转运rna(trna)、核糖体rna(rrna)、小核仁rna(snorna)、微小rna(mirna)、小干扰rna(sirna)、小核rna(snrna)及与piwi相互作用的rna(pirna)(amaraletal.,2008;martens-uzunovaetal.,2013)。mirna通过与靶mrna结合而起到翻译阻遏物的作用,其中,mirna在有足够的序列互补性的位点与靶mrna结合(ameresetal.,2007),而高度丰富的细胞质yrna通过影响ro蛋白的亚细胞定位而在rna质量控制中起作用(simetal.,2009)。除了在确定的亚细胞定位处沉默逆转录转座子的pirna外,其它类型的ncrna(包括sirna和内源sirna(endo-sirna))都有成熟mirna对mrna翻译的抑制活性(chuma和pillai,2009)。mirna活性依赖于在细胞质中的足够的丰度水平,以及与位于核内体膜(endosomalmembrane)的rna诱导的沉默复合物(risc)的相互作用(gibbingsetal.,2009;leeetal.,2009a),而低丰度的mirna对翻译抑制的影响较小。因此,某一mirna水平的细微变化可能已经影响了细胞过程,而强烈的扰动可以引起疾病。除丰度外,与(risc)蛋白质和rna伴侣的相互作用及正确的亚细胞定位也是控制mirna生理机能的相关因素(mullokandovetal.,2012;weeetal.,2012)。
mirna是一类小的、22~25个核苷酸的非编码调节rna,其以高度特异性的方式控制翻译后基因调节和功能的关键方面。由于mirna起特异性基因调节因子的作用,并且因为它们的表达在癌症发展中经常受到干扰,所以对其作为生物标记物的用途进行了研究。某一mirna(oncomir)的过表达(例如mir-21)或表达缺乏(例如mir200家族)似乎与临床上的侵袭性或转移性疾病结果相关。在慢性淋巴细胞性白血病(cll)中,循环的mirna已用于疾病归类及对治疗性干预响应的预测。
然而,虽然对mirna的图谱分析是相对标准的技术,但由于相互矛盾的数据,循环的mirna在临床上的广泛实施受到了限制。故需要能更好地预测个体健康状况的ncrna生物标记物。
技术实现要素:
本发明的一个方面提供了用于鉴定小非编码rna(ncrna)生物标记物对的方法,所述方法包括:
-对于ncrna,确定在第一体液样品中和在第二体液样品中的所述ncrna的两个不同种类的比例,所述第一体液样品获得自第一个体参照,所述第二体液样品获得自第二个体参照,其中,所述第一个体参照相对第二个体参照具有改变的健康状况,
-比较所述第一体液样品的比例和所述第二体液样品的比例,以及
-当比例在所述第一体液样品与所述第二体液样品之间发生改变时,鉴定所述ncrna的两个不同种类为生物标记物对,
其中,第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,并且第二种类是具有3'非模板核苷酸添加的ncrna,以及
其中,当所述ncrna是mirna时,所述第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切,并且所述第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切。
或者说,其中,第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,并且第二种类是具有3'非模板核苷酸添加的ncrna。
优选的,所述第一种类和所述第二种类选自典型的ncrna;在5'或3'末端剪切的和/或在5'或3'末端延伸的ncrna;和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切或者在5'或3'末端延伸;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,并且第二种类是具有3'非模板核苷酸添加的ncrna。
优选的,所述方法包括确定在第一体液样品中的所述ncrna的两个不同种类的量,并且确定在第二体液样品中的所述ncrna的两个不同种类的量。
优选的,第一个体参照来自一个或多个健康个体,并且所述第二个体参照来自一个或多个具有病症的个体,其中,所述病症优选优选为前列腺癌。
优选的,第一个体参照来自具有病症的个体,并且所述第二个体参照来自治疗所述病症后的同一个体。
在本文公开的方法的优选方面中,通过对各样品中的两个不同种类进行定量并确定两个量之间的关系,来确定生物标记物对的比例。优选的,使用深度测序(rna-seq)对上述种类进行定量。
本发明的另一方面提供了一种收集数据的方法,用于使用小非编码rna(ncrna)生物标记物对对个体的健康状况进行分类。该方法包括确定来自个体的体液样本中的生物标记物对的比例,将该比例与来自参照样品(例如上文所述的第二个体参照)的体液中的生物标记物对的比例进行比较,
其中,所述个体的比例和参照样品的比例之间是否存在差异有助于对所述个体的健康状况进行分类,
其中,所述生物标记物对由ncrna的两个不同种类组成,
其中,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,第二种类是具有3'非模板核苷酸添加的ncrna,以及
其中,当所述ncrna是mirna时,所述第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切,所述第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切。
或者说,其中,第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,第二种类是具有3'非模板核苷酸添加的ncrna。
优选的,所述第一种类和所述第二种类选自典型的ncrna;在5'或3'末端剪切和/或在5'或3'末端延伸的ncrna;和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切或者在5'或3'末端延伸;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,第二种类是具有3'非模板核苷酸添加的ncrna。
该方法中获得的数据可以单独或与其他因素(例如,额外的生物标记物的存在、患者的临床症状等)组合,来诊断个体具有病症。
本发明的另一个方面提供了使用小非编码rna(ncrna)生物标记物对,来对个体的健康状况分类的方法,所述方法包括确定来自所述个体的体液样本中的生物标记物对的比例,以及将该比例与来自参照样品的体液中的生物标记物对的比例进行比较,
其中,根据所述个体的比例和参照样品的比例之间是否存在差异,对个体的健康状况进行分类,
其中,所述生物标记物对由两个不同种类的ncrna组成,
其中,第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,第二种类是具有3'非模板核苷酸添加的ncrna,以及
其中,当所述ncrna是mirna时,所述第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切,所述第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切。
或者说,其中,所述第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,所述第二种类选自在5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,该具有3'非模板核苷酸添加的ncrna任选是在5'或3'末端剪切的;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,所述第二种类是具有3'非模板核苷酸添加的ncrna。
优选的,所述第一种类和所述第二种类选自典型的ncrna;在5'或3'末端剪切的和/或在5'或3'末端延伸的ncrna;和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切或在5'或3'末端延伸;
其中,当所述ncrna不是mirna时,所述第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,所述第二种类是具有3'非模板核苷酸添加的ncrna。
优选的,所述方法包括确定所述个体样品中的所述生物标记物对的量。
优选的,所述参照样品来自一个或多个健康个体。
优选的,所述参照样品来自一个或多个具有病症的个体。
优选的,本文公开的病症是癌症,更优选是前列腺癌、乳腺癌、chl、睾丸癌或结直肠癌。优选的,所述病症是前列腺癌、乳腺癌或chl。优选的,所述病症是阿尔茨海默氏病。
优选的,所述参照样品来自一个或多个个体,所述个体对病症治疗的响应良好或响应不良。
在本文公开的方法的优选方面,ncrna选自转运rna(trna)、核糖体rna(rrna)、snorna、微小rna(mirna)、sirna、小核(snrna)、yrna、穹窿体rna(vaultrna)、反义rna和piwirna(pirna),其中,所述ncrna优选选自mirna。
在本文公开的方法的优选方面,所述体液是尿液或血液。
在本文公开的方法的优选方面,两个不同种类选自典型的和3'nta-a;典型的和3'nta-g;典型的和3'nta-c;典型的和3'nta-u;3'nta-g和3'nta-c;3'nta-g和3'nta-u;3'nta-g和3'nta-a;3'nta-c和3'nta-u;3'nta-c和3'nta-a;及3'nta-u和3'nta-a;典型的和5'剪切的;典型的和3'剪切的;5'剪切的和3'nta-c;5'剪切的和3'nta-u;5'剪切的和3'nta-a;5'剪切的和3'nta-g;3'剪切的和3'nta-c;3'剪切的和3'nta-u;3'剪切的和3'nta-a;3'剪切的和3'nta-g;3'剪切的和5'剪切的;延伸的和3'nta-a;延伸的和3'nta-g;延伸的和3'nta-c;延伸的和3'nta-u;延伸的和5'剪切的;延伸的和3'剪切的;5'或3'剪切的和5'或3'延伸的;(剪切的和/或延伸的)和3'nta-u;(剪切的和/或延伸的)和3'nta-a;(剪切的和/或延伸的)和3'nta-g;(剪切的和/或延伸的)和3'nta-c;(剪切的和/或延伸的)和典型的。
在本文公开的方法的优选方面,所述生物标记物对的每一种ncrna都包括至少16个核苷酸。
图12提供了本发明的生物标记物对的示例性实施方式。表1理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的3'nta-a种类,例如典型的hsa-let-7g-5p和3'nta-a的hsa-let-7g-5p。因此,表1a公开了9组独立的生物标记物对。表1b公开了5组独立的生物标记物对(例如,典型的hsa-mir-200c-3p和3'nta-u的hsa-mir-200c-3p);表1c公开了4组独立的生物标记物对(例如,典型的hsa-mir-204-5p和3'nta-c的hsa-mir-204-5p);表1d公开了9组独立的生物标记物对(例如,典型的hsa-let-7f-5p和3'nta-g的hsa-let-7f-5p);表1e公开了11组独立的生物标记物对(例如,典型的hsa-let-7f-5p和3'剪切的hsa-let-7f-5p;表1f公开了3组独立的生物标记物对(例如,典型的hsa-mir-181b-5p和5'剪切的hsa-mir-181b-5p)。
在本文公开的方法的优选方面,生物标记物对选自图12中描述的生物标记物对之一。优选的,生物标记物对选自表1a。优选的,生物标记物对选自表1b。优选的,生物标记物对选自表1c。优选的,生物标记物对选自表1d。优选的,生物标记物对选自表1e。优选的,生物标记物对选自表1f。
图21提供了本发明的生物标记物的进一步示例性的实施方式。表2描述了用于表征前列腺癌的trna生物标记物对。表2理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。特别地,这些生物标记物对在确定尿液中的生物标记物对的方法中是有用的。表4和表10描述了可用于表征chl的mirna生物标记物对。特别地,这些生物标记物在确定从血液提取的胞外体囊泡(exosomalvesicle)中的生物标记物对的方法中是有用的。表4和10理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。表5和表11描述了可用于表征chl的mirna生物标记物对。特别地,这些生物标记物对在确定从血液提取的蛋白质组分中的生物标记物对的方法中是有用的。表5和表11理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。表6描述了可用于表征乳腺癌的mirna生物标记物对。特别地,这些生物标记物对在确定血液样品中的生物标记物对的方法中是有用的。表6理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。表7描述了可用于表征睾丸生殖细胞肿瘤的mirna生物标记物对。表7理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。表8描述了可用于结直肠癌的mirna生物标记物对。表8理解为公开了一种生物标记物是典型的序列,而另一种生物标记物是各自的变体。表9描述了可用于阿尔茨海默氏病的mirna生物标记物对。表9理解为公开了一个生物标记物是典型的序列,而另一个生物标记物是各自的变体。特别地,这些生物标记物对在确定血液样品中的生物标记物对的方法中是有用的。
在本文公开的方法的优选方面,生物标记物对选自表1、表2或表4~表11,优选选自表1、表2、表4、表5、表6和表9~表11。
优选的,在本文公开的方法中,由纯化自体液样品的胞外体囊泡,确定所述ncrna的两个不同种类的量。优选的,在本文公开的方法中,由纯化自体液样品的蛋白质组分,确定所述ncrna的两个不同种类的量。优选的,体液样品通过尺寸排阻色谱(sec)来纯化胞外体囊泡或蛋白质组分。
在本发明的另一方面,提供了一种用于表征个体中的典型霍奇金淋巴瘤(chl)的状况的方法,所述方法包括确定来自所述个体的体液样品中的一种或多种mirna的量,以及将所述一种或多种mirna的量与参照样品进行比较,其中,所述一种或多种mirna的存在或量的差异表征个体的状况。还提供了一种收集个体健康状况相关的数据的方法,包括确定来自所述个体的体液样品的一种或多种mirna的量,以及将所述一种或多种mirna的量与参照样品进行比较。该数据可用于表征个体中的典型霍奇金淋巴瘤(chl)的状况。
用于这些方法的一种或多种mirna选自表3中。
优选的,所述一种或多种mirna选自mir21-5p、let7a-5p、mir127-3p和mir155-5p。
表3描述了可用于表征个体中的典型霍奇金淋巴瘤(chl)的状况的mirna。特别地,这些生物标记物在确定从血液提取的蛋白质组分中的生物标记物对的方法中是有用的。优选的,该方法通过确定个体是否患有chl来表征chl的状况。
优选的,所述参照样品来自一个或多个健康个体。
优选的,所述参照样品来自一个或多个具有chl的个体。
优选的,表征chl的状况包括确定在接受chl治疗的个体中的治疗效果。优选的,所述参照样品来自接受chl治疗前的同一个体。
优选的,表征chl的状况包括确定患有chl的个体的预后。优选的,所述参照样品来自对疾病治疗的响应良好的一个或多个个体,或者来自对疾病治疗的响应不良的一个或多个个体。
在优选的实施方式中,所述方法进一步包括从体液样品纯化胞外体囊泡,以及确定与纯化的胞外体囊泡相关的一种或多种mirna的量。在优选的实施方式中,所述方法进一步包括从体液样品纯化蛋白质组分,以及确定与纯化的胞外体囊泡相关的一种或多种mirna的量。优选的,所述纯化包括,通过尺寸排阻色谱(sec)对体液样品进行处理。优选的,通过获得空隙体积分数来分离胞外体囊泡。
在优选的实施方式中,所述体液是血液。
优选的,本文公开的方法是在体外进行的,特别是量化相关生物标记物的步骤是在体外进行的。
附图说明
图1.来自b细胞及其胞外体的小rna全部组成。
a)样品特征和rna-seq数据的总结。用细胞和胞外体的小rna组分构建cdna文库。明确说明在定位至目标基因组(hsg19;人类基因组)之前和之后的所有文库的读序总数。为了注释定位至人类基因组的rna元件,针对目前已知的数据库分析所有定位的读序:1)来自mirbase版本19的成熟mirna和mirna前体的序列,2)于2013年2月6日下载的ncbi参照序列人refseq基因,3)来自基因组trna数据库的trna序列,4)利用从ucsc下载的repeatmasker算法检测的重复衍生序列,和5)从ncbi核苷酸数据库下载的piwirna(pirna)。
b)用细胞和胞外体的小rna组分构建cdna文库。注明了用于生成rna测序文库的样品组、细胞系和样品组分的类型。
c)来自细胞和胞外体组分的所有定位的读序通过对细胞转录物的注释进行分组,并且表示为定位的读序的分布频率(%),其中所有定位的读序都来源于细胞转录物。
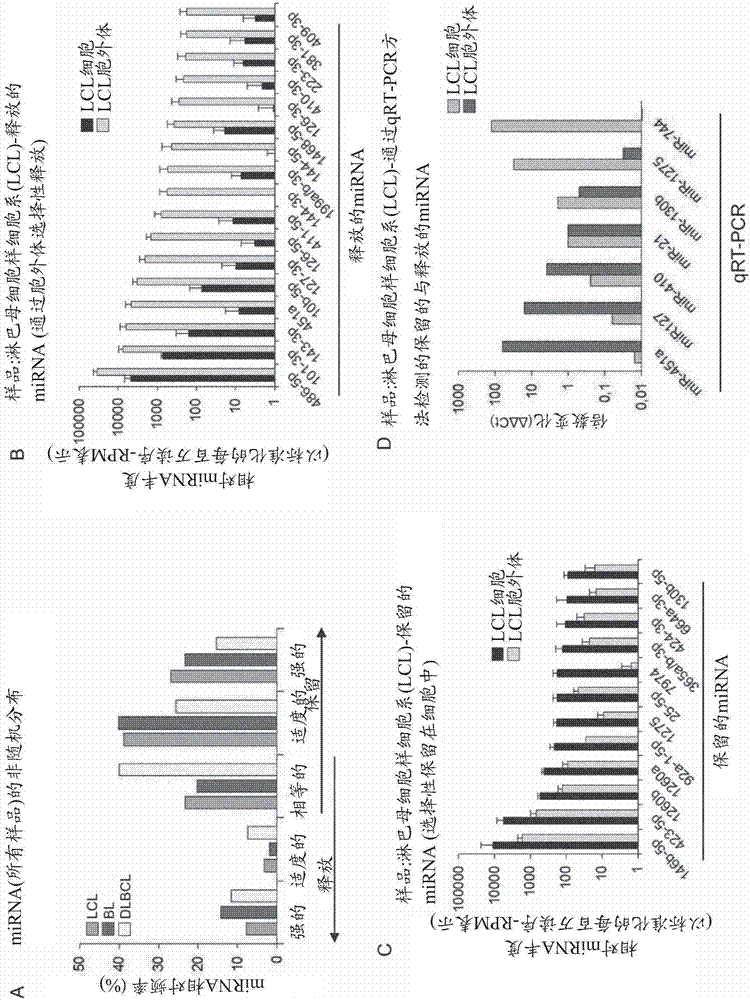
图2.微小rna非随机地掺入胞外体中。
a)根据标准化(每百万的读序,rpm)后的从细胞释放的mirna的量与保留在细胞中的量之间的比例,将mirna分为x轴表示的五组。将数据绘制为分布在五组中的mirna组分的百分比。
b~c)x-轴上描绘了在胞外体和细胞中显著过多出现的mirna,在y-轴(对数标度)描绘了它们的超过100读序/百万(标准化,rpm)的读序丰度。误差棒是平均值的sd(lcl1~3样品;n=3)。
d)通过基于颈-环的qrt-pcr在lcl细胞及其配对的胞外体中进行的mirna检测。数据表示相对于两次独立实验的mir-92act值进行标准化的技术重复的δδct值。
图3.微小rna非随机地掺入伯基特淋巴瘤胞外体中。
作为lcl样品,分析来自bl(bl1和bl2)的胞外体和细胞组分的mirna。edger分析产生在(3a)胞外体和(3b)细胞中统计学上显著过多出现的mirna,它们的读序丰度超过每百万100个读序(标准化,rpm)。y轴表示非随机分布的mirna的相对丰度。将显著被排出或保留的mirna与其被保留或排出的等同物进行比较。误差棒是sd;(n=2);y轴以对数标度表示。
图4.ebv编码的mirna显示出比人mirna更强的细胞保留趋势。
a)根据标准化(rpm)后的从细胞释放的mirna的量与保留在细胞中的量之间的比例,将人(lcl)和ebv的mirna分为五组。数据绘制为在五组中分布的mirna组分的百分比。
b)根据标准化(rpm)后的从细胞释放的mirna和保留在细胞中的mirna之间的倍数变化(定义为胞外体/细胞log2)来对人(lcl1)和ebv的mirna分类。垂直线表示细胞保留或胞外体相关释放的增加>4倍的mirna组分。红线表示病毒mirna在人mirna(灰色)中的分布。
图5.在各lcl样品(lcl1、2和3;细胞对胞外体)中成熟mirna及其亚型的分布。a)将在每一文库中检测到的定位至注释的mirna序列的所有mirna的测序读序进一步解析为成熟(典型的)序列和亚型序列。它们进一步细分为转录后修饰的亚型(非模板核苷酸添加;nta)和转录后未修饰的亚型(截短和延伸)。b)样品针对分配于每一样品的各mirna所有读序的总和(成熟与亚型)的百分比来作图,和c)针对具有nta的所有mirna读序的总和的百分比来作图。
图6.3'-末端nta型对mirna变体在细胞和胞外体之间分布的影响的统计分析和显著性。对在所有12个样品中表达的118种mirna的伪发现率校正的p值。来源于回归模型的p值,对细胞和胞外体之间的各nta(a,u,c和g)的计数比例进行比较。在每个图中,圆圈颜色指示效应的方向:胞外体>细胞意味着数据表明与细胞相比,胞外体中的比例更大,而胞外体<细胞意味着数据表明与胞外体相比,细胞中的比例更大。结果显示3'末端腺苷酸化(adenylation)的mirna亚型优先被保留(卡方检验,p<0.05),而所有其他nta优先被释放(3'末端u:卡方检验,p=0.02;3'末端c:费舍尔(fisher)精确检验,p=0.04;和3'末端g:fisher精确检验,p=0.02)。
图7.3'末端转录后修饰界定了mirna的保留或释放。
a~b)比较lcl样品中成熟mirna及其nta修饰或未修饰的亚型在细胞和胞外体之间的分布。误差棒是平均值的sd(lcl1~3样品,n=3);*p值=0.005(学生t检验)。
c)各lcl细胞和胞外体库之间的mir-486-5p的分布。将定位至mir-486-5p的所有测序读序相加,并且mir-486-5p的(亚)型的组成以百分比表示。
d)通过qpcr检测细胞(lcl)和胞外体rna组分中的典型mir-486-5p、3'-末端腺苷酸化的mir-486-5p(3'-aaa)和3'末端尿苷化的mir-486-5p(3'-uuu)。数据表示来自两次独立实验的技术重复的平均循环阈值(ct)值(误差棒,sd)。
图8.3'末端腺苷酸化的程度使保留增加,而3'末端尿苷化的程度区分胞外体小rna负荷。
a和d)3'-a和3'-u亚型的成对分析的频率图。选择在细胞和胞外体中均发现表达的mirna(>300个读序,100个mirna)来分析它们的腺苷酸化和尿苷化读序的分布,以占全部nta修饰读序的百分比来表示。红色圆圈:在胞外体中具有较高百分比的亚型;紫色圆圈:在细胞中具有较高百分比的亚型;白色圆圈:均匀分布。
b~c)人mirna和病毒mirna的组分,对其腺苷酸化读序(3'末端-a)显示出保留或释放的趋势,这是针对相同mirna的非腺苷酸化读序测量。腺苷酸化的mirna具有更高的保留概率,并且随着添加的腺嘌呤数目而增加。
e~f)相比于3’末端腺苷酸化mirna亚型,3'末端尿苷化mirna亚型更常存在于lcl胞外体、bl胞外体和人尿液胞外体中。柱子表示具有3'末端nta的每个样品(仅胞外体)亚型读序的加权平均值;核苷酸类型在x轴上表示。误差棒是3个lcl样品的sd(n=3);p=0.005;2个bl样品(n=2);p=0.001;和6个来自人尿液的样品(n=6);p<0.0001(学生t检验)。
图9.3'-末端转录后修饰影响加工的小ncrna的分布。a)具有3'末端腺苷酸化或b)3'-末端尿苷化(右图)的yrna片段在细胞和胞外体组分间的差异化分布。对应于加工的yrna片段的rna部分针对具有3'末端修饰的rna片段的百分比作图,其中加工的yrna片段源自rny1、rny3、rny4和rny5(x-轴)。
图10.通过小rna测序在尿液胞外体中鉴定的定位的读序列表。人尿液细胞外囊泡的样品特征和rna-seq数据的总结。在纯化尿液胞外体后,由胞外体小rna组分产生cdna文库。确定了定位至目标基因组(hsg19/grch37,补丁5)之前和之后的所有文库的读序总数。为了向定位至人类基因组的rna元件提供注释,针对当前已知的数据库分析所有定位的读序:1)来自mirbase第19版的成熟的和mirna前体序列,2)2013年5月下载的ncbi参照序列人refseq基因,3)来自基因组trna数据库的trna序列,4)利用从ucsc下载的repeatmasker算法检测的重复衍生序列,和5)从ncbi核苷酸数据库下载的piwirna(pirna)。
图11.健康肾组织活检与明确的细胞肾细胞癌(肾组织)活检
a)微小rna多样化。每个库中检测到的定位至注释的mirna序列的测序读序被分组并且表示为一个mirna。这些定位的读序由成熟的(典型的)和亚型序列组成。亚型进一步细分为酶修饰的亚型(3'末端nta非模板核苷酸添加)和未修饰的亚型(在3'-末端和5'-末端发生截短和延伸)。
b)通过rnaseq对来自明确的细胞肾细胞癌(ccrc;4个实验组)的公众可获得的数据进行分析。基于成熟(mirbase)序列的数据分析显示所选择的mirna在实验组之间难以分辨。
c)所有4个实验组中,mirna亚型和成熟mirna之间的比例显示3'-端腺嘌呤添加(腺苷酸化)(特别是临床相关组之间)具有显著差异。腺苷酸化mirna的比例的显著差异能区分开临床定义的组。
图12.转录后修饰对健康与前列腺癌患者进行分级。尽管该表示出数据表示为“c/v”,但是该比例实际上表示“v/c”。此更改不会影响std或p值。
图13.用于监测恶性淋巴瘤患者中的重要肿瘤组织的非侵入性策略
a)左:治疗前典型的霍奇金淋巴瘤患者的fdg/pet影像。黑色箭头表示多个代谢活跃的肿瘤块。右:同一患者的融合的pet/ct影像。白色箭头表示重要的肿瘤块。b)淋巴瘤肿瘤细胞(上部深棕色)和正常细胞(下部浅棕色)主动地分泌100纳米囊泡,包括进入循环的来源于mvb的胞外体。这些细胞外囊泡(ev)含有免受外部rna酶影响的mirna。除了由ev包裹,mirna可以与蛋白质和hdl相关联并且经蛋白质和hdl保护而免于降解,然而这些不反映重要的肿瘤细胞。此外,死亡细胞将生物分子(即rna,dna,蛋白质)释放到循环中。该图改编自horietaletal.,2013sciencetranslmed。
图14.为了对在患者血浆中的mirna检测进行优化,通过尺寸排阻色谱(sec)一步将循环的细胞外囊泡(ev)与蛋白质/hdl中分离
a)qev尺寸排阻色谱(sec)柱(近期从izontm购得)。qev柱允许循环的ev从1~1.5ml血浆中一步可再生地分离,将它们与循环的蛋白/hdl分离(boingetal.,jev2014)。b)使用琼脂糖cl-2bsec从chl患者分离的血浆ev的em图像。尺寸条表示200nm,并且虽然存在更大(200nm)的囊泡,大多数ev在100nm的范围内。c)与b中相同的患者血浆的蛋白质/hdl组分的em图像。未观察到类似于ev的颗粒。d)基于可调电阻性脉冲感测(trps)使用qnano(izontm)的颗粒分析。使用qev装置分离chl患者血浆ev和蛋白/hdl组分。ev组分高度富集于对应于b中em图像的尺寸分布(橙色)的颗粒中。e)通过sec分离的ev和蛋白质/hdl组分的rt-pcr分析。ev组分9-12中vtrna1-1是高度富集的,而蛋白质/hdl组分(19-22)中富集了mir92a,与(arroyoetal.,pnas2011)相一致。
图15.血浆ev中候选mirna生物标记物的选择和验证
a)来自囊泡的mirna的分级聚类。来自chl患者(治疗前)和健康对照的淋巴瘤细胞系衍生的胞外体(n=7)和血浆细胞外囊泡的rnaseq分析显示明确的不同的图谱。b)血小板、pbmc和ev中10种最丰富的mirna列表(pleetal.,plosone2012)。c)血浆ev和霍奇金(hodgkin)细胞系衍生的胞外体(l1236)中mirna的水平的比较产生潜在的chl基质衍生的和肿瘤相关的mirna标志物。mirna通过log(fc)>1显示区别。d)箱线图显示了从chl患者(n=10)和健康对照(n=4)中分离的血浆ev中的4种候选mirna生物标记物的rt-pcr数据。在来自chl患者和健康对照的血浆ev中均检测到大量mir1973。使用双尾学生t检验计算p值。
图16.成功治疗期间,血浆ev中的mir-21-5p和let7a-5p水平降低
a)在两轮一线治疗(beacopp)之前(左)和之后(右),chl患者的pet影像。箭头表示代谢活跃的肿瘤。b)rt-pcr分析显示在治疗期间ev相关的mir-21-5p和let-7a-5p强烈降低(70%)。使用mir-1973标准化mir-21-5p和let-7a-5p水平并显示为倍数降低(误差棒,sem)。c+d)经rt-pcr确定,在beacopp治疗(c)期间的初治患者和在用dhap/adc治疗,随后进行自体干细胞移植的复发患者(d)中,ev中的mir-21-5p和let7a-5p水平降低。使用mir-1973标准化mir-21-5p和let-7a-5p水平并显示为倍数降低(误差棒,sem)。e+f)通过降低血清tarc水平(e)确定的,响应治疗的chl患者中ev降低的mir-21-5p和let-7a-5p水平,而在chl患者中不降低(其中tarc水平保持高水平)(f)。使用mir-1973标准化mir-21-5p和let-7a-5p水平并显示为倍数变化(误差棒,sem)。
图17.在随访期间候选mirna生物标记物在血浆ev中保持低水平
a)在beacopp治疗期间(t=1个月)和之后(t=3个月)的中,chl患者血浆ev中的mir-127-3p、mir-155-5p、mir-21-5p和let-7a-5p水平降低,并在治疗后6个月内保持低水平。使用mir-1973标准化mirna水平并显示为倍数降低(误差棒,sem)。b)rt-pcr分析显示,健康供体血浆ev中的mir-21-5p和let-7a-5p水平是稳定的。数据显示为ct值(误差棒,sem)。
图18:chl血浆ev中的候选mirna的水平升高是疾病特异性的
与健康对照(n=4)相比,mir-127-3p、mir-155-5p、mir-21-5p和let-7a-5p的rt-pcr水平在chl患者血浆ev(n=10)中增加,但在自身免疫(sle)患者中(n=3)不增加。数据显示为ct值(误差棒,sem)。使用双尾学生t检验计算p值。
图19:健康的和霍奇金患者的血浆ev中mirna的nta分析
我们对来自健康供体和chl患者的多个血浆的蛋白质和囊泡组分中进行测序,并用srnabench(http://arn.ugr.es/srnabench/)分析数据。a)显示所有鉴定为3'-末端nta定义为a或u的mirna比例图。只有chlev组分具有更高比例的3'-末端尿苷化mirna。b)与a相同,但现在数据表示基于3'末端转录后nta的修饰的mir-486-5p的差异。c)2个供体的各种组分中mir486-5p的taqmanrt-pcr,其证实了如b所示rnaseq数据,taqmanrt-pcr如前所述的(koppers-lalicetal.,2014)。
图20:与mir-92a基因组序列比对的匹配读序的图示。5'或3'-末端核苷酸的转录后修饰(nta)和伸长/截短用加下划线的字母(用于延伸)或*表示截短。
图21:示例性生物标记物。表2描述了来自前列腺癌(pca)患者尿液的trna生物标记物对。表3描述了表征chl的mirna。表4和表10描述了从健康供体和chl患者的血浆中提取的细胞外囊泡中的ncrna生物标记物对。表5和表11描述了从健康供体和chl患者的血浆中提取的蛋白质组分(pf)中的ncrna生物标记物对。表6描述了乳腺癌患者血清中的ncrna生物标记物对。表7描述了睾丸生殖细胞肿瘤中的ncrna生物标记物对。表8描述了在结直肠癌中的ncrna生物标记物对。表9描述了阿尔茨海默病患者血清中的ncrna生物标记物对。
具体实施方式
如本文所使用的,冠词“一/一个/一种(a)”和“一/一个/一种(an)”指该冠词语法上修饰的对象的一个或一个以上(即至少一个)。作为示例,“一个元件(anelement)”是指一个元件或一个以上元件。
如本文所使用的,“体液”是指含ncrna的体液,其包括血液(或血液组分,例如血浆或血清)、淋巴液、粘液、液泪、唾液、痰、尿液、精液、粪便、csf(脑脊液)、母乳和腹水。优选的,所述体液是尿液。优选的,所述体液选自血液。如本文所使用的,“血液”还包括血清和血浆。优选的,所述体液是血浆。
“生物标记物”可用于指个体中以不同浓度存在的,可用于预测个体健康状况的生物分子。
如本文所使用的,“包括/包含”及其变化形式以其非限制性意义使用,意指包括该词之后的项目,但不排除未具体提及的项目。此外,动词“由……组成”可被替换成“基本上由……组成”,意味着本文定义的化合物或辅助化合物可以包括除了具体指出的那些之外的额外组分,所述另外的组分不改变本发明独特的特征。
如本文所使用的,“健康状况”是指个体在特定时间的整体(身体/生理)症状。健康状况包括是否存在疾病或病症,例如神经病症(例如阿尔茨海默氏病)、癌症/肿瘤、感染性疾病、代谢性疾病(例如淀粉样变性)、心血管疾病和免疫性病症。优选的,所述病症选自前列腺癌、乳腺癌、子宫颈癌、淋巴瘤、结肠癌、成胶质细胞瘤和肺癌。
健康状况还包括发生此类病症的风险,即,一般群体(或具有相似的年龄、遗传背景、环境风险因素等的个体)具有降低或增加的风险。健康状况还包括疾病/病症的特定阶段,及严重性和预后(例如,存活预后)。健康状况还指要经特定药剂有效治疗的个体的预后。“对健康状况分类”包括将个体分类为健康的;具有或不具有特定病症;发生病症的风险增加、降低或是正常;对特定治疗的响应良好者或响应不良者;具有特定阶段或严重性的疾病;生存或恢复预后良好或不良。
如本文所使用的,“个体”是指人和动物,例如哺乳动物,如家畜(例如狗、猫);农场动物(例如牛、绵羊、猪、马);或实验室动物(例如猴、大鼠、小鼠、兔、豚鼠)。优选的,所述个体是人。
如本文所使用的,“小非编码rna”(ncrna)是指不翻译成蛋白质的rna,且其包括转运rna(trna)、核糖体rna(rrna),snorna、微小rna(mirna)、sirna、小核(snrna)、yrna、穹窿体rna、反义rna、tirna(转录起始rna)、tssa-rna(转录起始位点相关rna)和piwirna(pirna)。小ncrna的长度小于200个核苷酸。
优选的,所述ncrna是小poliiirna,优选选自trna、mirna、snrna、yrna,穹窿体rna和snrna。优选的,所述ncrna选自mirna、yrna、穹窿体rna,更优选的,所述ncrna是mirna。
优选的,本文所用的小ncrna长度为16~200个核苷酸,更优选16~100个核苷酸,甚至更优选16~40个核苷酸。ncrna可以是内源性来源(例如,人mirna)或外源性来源(例如,病毒、细菌、寄生虫)。
“典型的”ncrna是指从基因组序列预测的rna序列,并且是被鉴定为特定rna的最丰富的序列。对于mirna来说,这是指通过“典型的mirna通路”形成的mirna。mirna前体(pre-mirna)被切割成短发夹rna,然后输出到细胞质中,以通过核糖核酸内切酶(dicer酶)进行加工。所得到的“典型的”成熟mirna的长度通常为21~22个核苷酸。
“剪切的”ncrna是指其中已经经由核酸外切酶介导的核苷酸剪切,去除了分子5'末端和/或3'末端的一个或多个核苷酸的ncrna。优选的,所述剪切是3'剪切。优选的,从典型序列的3'末端剪切一个核苷酸。由于典型的mirna的起始位点和终止位点是已知,所以可以容易地检测到剪切的mirna。在图20和图21中(参见例如表11c)描述了剪切的ncrna的实例。
3'非模板核苷酸添加(3'nta)是指通常通过rna核苷酸转移酶,例如papd4、papd5、zcchc6和zcchc11,翻译后在rna的3'末端添加一个或多个核苷酸(burroughsetal.,2010;polikepahadandcorry,2013)。最常见的形式是腺苷酸化(3'nta-a)和尿苷化(3'nta-u),但是也可添加胞嘧啶(3'nta-c)和鸟嘌呤(3'nta-g)。虽然通常添加一个或两个核苷酸,但也可添加三个或更多个核苷酸。在nta发生之前,典型的序列任选地在5'末端或3'末端剪切。优选的,3'nta是选自3'nta-a、3'nta-g、3'nta-u和3'nta-c的单核苷酸添加。在图20和图21中(参见例如表2a)描述了3'ntancrna的实例。
“延伸的ncrna”是指比典型的mirna序列长的mirna,并且是本领域中公认的术语。构成延伸的核苷酸对应于前体序列的核苷酸,因此与非模板核苷酸添加不同,构成延伸的核苷酸是由基因组编码的。不希望被理论所束缚,认为延伸的ncrna是mirna前体差异化加工的结果。通常来说,与典型的mirna序列相比,该延伸可以包括1~5个,通常1~3个额外的核苷酸。由于典型的mirna的起始位点和终止位点是已知,所以可以容易地检测到延伸的mirna。在图20和图21中(参见例如表11a)描述了延伸的ncrna的实例。
ncrna变体选自ncrna的典型序列,ncrna的5'或3'剪切形式,ncrna的5'或3'延伸形式,ncrna的5'或3'剪切形式或ncrna5'或3'延伸形式(本文中统称为“长度变体”),和具有3'非模板核苷酸添加(3'nta)的ncrna。如本文所使用的,“ncrna变体”是指源自一个ncrna基因的一组序列。每个变体例如通过5'或3'剪切,或存在或不存在3'nta,而在序列上彼此不同。变体可具有相同或不同的生物学功能。
不希望被理论所束缚,认为剪切的ncrna(即截短的)和延伸的ncrna(即延长的)在pre-mirna加工中可导致相似的低效率。在一些实施方式中,ncrna变体是“长度变体”,即剪切的或延伸的ncrna。例如,表5b列出了生物标记物对,其中一个变体是典型的结构,而第二变体是3'长度变体。在一些实施方式中,优选将长度变体分成延伸的ncrna和剪切的ncrna。表11是对相同数据的分析,但是其中长度变体被分成延伸的ncrna(参见对于3'末端延伸的表11a)和剪切的ncrna(参见对于3'末端剪切的变体的表11b)。
定量(quantify)和量化(quantification)可以互换使用,并且是指确定样品中的物质(例如,生物标记物)的(相对或绝对的)量或丰度的过程。例如,可以通过包括但不限于微阵列分析、qrt-pcr、rna印记(northernblot)上的条带强度、定向小rna测序的方法,或通过本领域已知的各种其它方法,来定量。
术语“治疗”包括预防性和/或治疗性治疗。术语“预防性或治疗性”治疗是本领域公知的,包括将一种或多种受试组合物给药予受体。如果在临床表现出不想要的症状(例如,受体动物的疾病或其他不想要的状况)之前给药,则治疗是预防性的(即,保护受体免于发生不想要的症状),而如果在表现出不想要的症状之后给药,则治疗是治疗性的(即,旨在减少、改善或稳定现有的不想要的症状或其副作用)。
一方面,本发明提供了一种鉴定小非编码rna(ncrna)生物标记物对的方法。优选的,ncrna生物标记物的长度至少为16个核苷酸。实施例3描述了用于将健康患者与前列腺癌患者区分开的生物标记物对,其使用本文所述的方法来鉴定。最好的生物标记物对公开于图12中。
上述方法包括确定ncrna在第一体液样品中和在第二体液样品中所述ncrna的两个不同种类的比例,所述第一体液样品获得自第一个体参照,所述第二体液样品获得自第二个体参照,其中,所述第一个体参照相对于第二个体参照健康状况发生改变。
所述体液样品来自一个或多个个体,或者可以是个体群体中的平均比例。所述第一个体参照(其可以是一个以上的个体,优选一个)相对于第二个个体参照(其可以是一个以上的个体,优选一个)健康状况发生改变。在一些实施方式中,第一个体参照和第二个体参照来自同一个体,其中个体的健康状况发生了改变,例如,样品获得自治疗方案之前和之后的个体。
诊断和预后生物标记物的鉴定方法是本领域公知的。样品的选择完全在本领域技术人员的范围内,并且可以根据待鉴定的生物标记物的类型而改变。
在疾病的生物标记物的情况下,示例性实施方式提供了来自具有病症的个体的第一个体参照,以及来自不具有所述病症的个体参照的第二样品。优选的,所述病症是癌症,更优选选自宫颈癌、淋巴瘤、结肠癌、成胶质细胞瘤和肺癌、前列腺癌。
在风险的生物标记物的情况下,示例性实施例提供了来自病症发生风险增加的个体的第一个体参照,以及来自风险未增加的个体的第二个体参照。例如,可由肺癌发病前的重度吸烟个体提供样品。分析比例,并且在一定的时间(例如1年或2年)后监测个体中的肺癌状况。
在预后的生物标记物的情况下,示例性实施例提供来自成功治疗的个体的第一个体参照,以及来自未成功治疗的个体的第二样品。
可通过技术人员已知的方法确定rna种类的比例。优选的,通过定量每一样品中的两个不同种类,并确定两个量之间的关系来确定所述比例。这优选表示为一个量除以另一个量的商。优选的,确定每一种类的丰度。更优选的,确定每一种类的相对丰度。
在一个示例性实施方式中,比例等于(ncrna种类1的丰度除以典型的ncrna的丰度)除以(ncrna种类2的丰度除以典型的ncrna的丰度)。
定量rna水平的方法是众所周知的。有几种方法向ncrna添加额外的序列,以便于引发和检测。特别地,可以向各ncrna添加共同序列,以允许使用单一的通用延伸引物。特别地,基于qpcr的ncrna检测方法向mirna添加额外的序列,以在逆转录之前或期间增加其长度。还存在许多用于对小rna进行pcr测定的市售系统(例如appliedbiosystemscustom
所述方法还包括比较来自所述第一体液样品的比例和所述第二体液样品的比例,并且当所述比例在所述第一体液样品和所述第二体液样品之间发生改变时,将所述ncrna的两个不同种类鉴定为生物标记物对。在第一样品和第二样品之间发生改变的比例将所述ncrna鉴定为ncrna生物标记物。更准确地说,两个ncrna种类被共同鉴定为生物标记物。改变的比例是两个样品的比例之间存在统计学的显著性差异。优选的,当来自第一样品的比例落在第二样品的约1.0标准偏差、约1.5标准偏差、约2.0标准偏差或约2.5标准偏差之外时,则鉴定为改变的比例。
ncrna种类选自典型的ncrna序列,5'或3'剪切形式的ncrna,5'或3'延伸形式的ncrna,5'或3'剪切形式的ncrna或5'或3'延伸形式的ncrna(即“长度变体”),和具有3'非模板核苷酸添加(3'nta)的ncrna。优选的,第一种类选自典型的ncrna,而第二种类是具有3'非模板核苷酸添加的ncrna,并且当所述ncrna是mirna时,第一种类选自典型的ncrna、在5'末端或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'末端或3'末端剪切,第二种类选自在5'末端或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'末端或3'末端剪切。清楚地是该比例是相同ncrna的两个不同种类的比例。
在示例性实施方式中,所述比例选自典型的:3'nta-a;典型的:3'nta-g;典型的:3'nta-c;典型的:3'nta-u;3'nta-g:3'nta-c;3'nta-g:3'nta-u;3'nta-g:3'nta-a;3'nta-c:3'nta-u;3'nta-c:3'nta-a;和3'nta-u:3'nta-a;典型的:5'剪切的;典型的:3'剪切的;5'剪切的:3'nta-c;5'剪切的:3'nta-u;5'剪切的:3'nta-a;5'剪切的:3'nta-g;3'剪切的:3'nta-c;3'剪切的:3'nta-u;3'剪切的:3'nta-a;3'剪切的:3'nta-g;3'剪切的:5'剪切的;延伸的和3'nta-a;延伸的和3'nta-g;延伸的和3'nta-c;延伸的和3'nta-u;延伸的和5'剪切的;延伸的和3'剪切的;5'或3'剪切的和5'或3'延伸的;(剪切的和延伸的)和3'nta-u;(剪切的和延伸的)和3'nta-a;(剪切的和延伸的)和3'nta-g;(剪切的和延伸的)和3'nta-c;(剪切的和延伸的)和典型的。优选的,所述比例选自典型的:3'nta,或3'nta:不同的3'nta。比例中的分子和分母可以颠倒对于技术人员来说是清楚的。优选的,ncrna是mirna。
在示例性实施方案中,所述比例选自典型的:3'nta-a;典型的:3'nta-g;典型的:3'nta-c;典型的:3'nta-u;3'nta-g:3'nta-c;3'nta-g:3'nta-u;3'nta-g:3'nta-a;3'nta-c:3'nta-u;3'nta-c:3'nta-a;和3'nta-u:3'nta-a。比例中的分子和分母可以颠倒对于技术人员来说是清楚的。
本发明证明了通过观察ncrna的两个种类的比例,可以改进ncrna生物标记物的预测能力。实施例2表明,四种不同的mirna的典型序列无法区分健康组织和肾癌。然而,当使用比例3'nta-a:典型的mirna时,可通过这四种相同的mirna来区分临床相关组。
因此,本发明还提供了使用小非编码rna(ncrna)生物标记物对,来区分个体的健康状况和收集关于个体的健康状况的数据的方法。ncrna对可以基于任何已知的ncrna生物标记物或者那些例如通过本文所述的方法鉴定的ncrna生物标记物。优选的,每一ncrna生物标记物的长度都为至少16个核苷酸。已经建议将各种ncrna分子(特别是)mirna用作生物标记物(参见例如,关于黑素瘤标记物的wo2009099905,关于肺病标记物的wo2011025919;关于阿尔茨海默病标记物的wo2013003350;关于气道疾病标记物的us2012289420等)。这些ncrna分子中的任何一种都可以用于上述方法的实施方式中。
用于对健康状况进行分类的方法包括,确定来自个体的体液样品中的生物标记物对的比例,将该比例与来自参照样品的体液中的生物标记物对的比例进行比较,其中,根据所述个体的比例与参照样品的比例之间是否存在差异(即,其中的比例发生改变)来区分所述个体的健康状况。
可从一个个体或从一群个体的平均值来获得的参照样品。参照样品的比例可通过本文所述方法来确定,或者它可以是先前获得的信息。与参照样品相比,所述个体的比例发生改变意味着与参照相比健康状况发生改变。
选择合适的参照样品在本领域技术人员的知识范围内。在一些实施方式中,参照样品来自具有特定病症的个体,而改变意味着所述个体没有患该病症。
在优选的实施方案中,来自个体的比例与两个参照样品进行比较,其中第一参照样品是“健康的”对照样品,第二参照样品来自健康状况(例如,处于发展特定疾病的风险)发生改变的个体。技术人员应认识到,当个体的比例更接近于来自“改变的健康状况”的比例时,则该个体的健康状况很可能也发生了改变。
所述生物标记物对由ncrna的两个不同种类组成,其中,第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,并且第二种类是具有3'非模板核苷酸添加的ncrna,其中,当所述ncrna是mirna时,第一种类选自典型的ncrna、在5'或3'末端剪切的ncrna,和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切;而第二种类选自5'或3'末端剪切的ncrna和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切。比例的确定和优选的rna种类如本文先前所述的。优选的,生物标记物对由ncrna的两个同种类组成,其中第一种类和第二种类选自典型的ncrna;在5'或3'末端剪切的ncrna和/或在5'或3'末端延伸的ncrna;和具有3'非模板核苷酸添加的ncrna,任选地在5'或3'末端剪切或在5'或3'末端延伸;
其中,当所述ncrna不是mirna时,第一种类选自典型的ncrna和具有3'非模板核苷酸添加的ncrna,而第二种类是具有3'非模板核苷酸添加的ncrna。
在优选的实施方式中,本文公开的方法确定了个体对进行的特定治疗做出响应的可能性。优选的,所述方法还包括治疗所述个体的所述疾病或病症的步骤。在优选的实施方式中,所述方法确定个体发生特定病症的风险。
在优选的实施方式中,本文公开的方法诊断患有疾病或病症的个体。在优选的实施方式中,所收集的数据可以用作辅助执业医师进行诊断或预后的一个因素。优选的,所述方法还包括治疗所述个体的所述疾病或病症的步骤。
在优选的实施方式中,所述病症是前列腺癌,并且生物标记物对选自表12(来自实施例3)或表2。在优选的实施方式中,所述病症是chl,并且生物标记物对选自表4或表5。在优选的实施方式中,所述病症是乳腺癌,并且生物标记物对选自表6。在优选的实施方式中,所述病症是阿尔茨海默氏病,并且生物标记物对选自表9。
本发明还提供了用于表征个体中典型霍奇金淋巴瘤(chl)的状况的方法,和收集关于个体的健康状况的数据的方法。所述方法包括确定所述个体的体液样品中的一种或多种mirna的量,并将所述一种或多种mirna的量与参照样品进行比较。可如本文已经公开的方法确定所述mirna的量。
实施例6描述了鉴定用于对chl进行分类的mirna。相应地,在这些方法中所使用的一种或多种mirna选自表3。优选的,一种或多种mirna选自let-7a-5p、mir-1908-5p、mir-5189-5p、mir-92b-3p、mir-425-3p、mir-625-3p、mir-7706、mir-125b-5p、mir-760、mir-21-5p、mir-122-5p,更优选的,选自mir-21-5p、let-7a-5p、mir-127-3p和mir-155-5p。
可能已经从单一个体或从个体群体的平均值获得了参照样品。
在一些实施方式中,参照样品来自一个或多个健康个体(即,未患有chl的个体)、一个或多个患有chl的个体、在接受chl治疗前的同一个体,对病症治疗的响应良好的一个或多个个体,或来自对病症治疗的响应不良的一个或多个个体。例如,所述一种或多种mirna的量与从健康或患病个体获得的参照水平相比,可用于表征个体是否患有chl。治疗后的所述一种或多种mirna的量与治疗前个体中的水平相比,可用于表征对治疗的响应,例如治疗是否有效或患者是否已复发。治疗后的所述一种或多种mirna的量与从已知对治疗的响应良好或不良的患者获得的参照水平相比,可用于表征个体治疗的预后是否良好。
在本发明的另一方面中,可以通过纯化体液(即,将体液分成不同的生化组分),来改善生物标记物/生物标记物对的鉴定,以及所述生物标记物/生物标记物对健康状况的表征。血液中的大多数细胞外ncrna与可溶性生化组分相关,其中可溶性生化组分包括蛋白质复合物、脂质囊泡(ldl和hdl)、细胞外囊泡。在优选的实施方式中,优选使用尺寸排阻色谱法富集体液,使体液富含细胞外囊泡(即胞外体囊泡)或富含高蛋白质(蛋白质/hdl)组分。图14e描述了使用尺寸排阻色谱法分离胞外体囊泡和高蛋白质组分,以及在这两种组分之间mirna分布的差异的实例。表4进一步公开了可区分健康供体和chl患者之间的血液中的胞外体囊泡组分的生物标记物对。表5公开了可区分健康供体和chl患者之间的血液中的蛋白质组分的生物标记物对。
本说明书中引用的所有专利和参考文献通过引用方式整体并入本文中。
通过以下实施例进一步解释本发明。这些实施例仅用于阐明本发明,而并不限制本发明的范围。
实施例
实施例1
eb病毒(ebv)-转化的淋巴母细胞b细胞(lcl)组成型分泌大量的内体来源的细胞外囊泡(ev),该细胞外囊泡(ev)被称为胞外体,其包含人和病毒的mirna。拷贝数测量表明胞外体介导mirna的细胞-细胞传递,导致了在受体细胞中的积累和靶mrna的抑制(pegteletal.,2010)。越来越多的体外和体内数据暗示,内体膜处的mirna分选支持了胞外体在细胞-细胞通讯中的功能(vanbalkometal.,2013;kosakaetal.,2010;mittelbrunnetal.,2011;montecalvoetal.,2012;pegteletal.,2010;umezuetal.,2013;zhangetal.,2010;zhuangetal.,2012)。值得注意的是,在内体膜处干扰胞外体的形成减少了功能性mirna的分泌(kosakaetal.,2013;mittelbrunnetal.,2011)。虽然mirna通过ev的分泌可能以非随机方式发生(bellinghametal.,2012;guduric-fuchsetal.,2012;nolte-’thoenetal.,2012;palmaetal.,2012),但对纯化的胞外体群和生产细胞一直缺乏全面的对比分析。
小非编码rna家族在b细胞和胞外体之间差异化分布。
为了确定小于200个核苷酸的小rna种类是否经历掺入胞外体的选择,我们生成了细胞和相应的胞外体的小rna文库,用于对ebv驱动的lcl(n=3)和三个淋巴瘤细胞系(n=3)进行测序。这一方式提供了全面的数据集,以在定义的细胞背景内对细胞内和细胞外的全部rna进行高性能的统计分析。rna-seq产生了每个样品>100万个基因组定位读序(mappedread)(图1中为各读序和比对统计),这些基因组定位读序与人和ebv基因组都进行了比对。与rna参照文库的比较揭示了细胞和胞外体的小rna组分含有来自不同类别的rna的产物。除了小rna序列的较大的多样性之外,几类rna似乎富集于细胞(图1;黑色柱)中,而其他的rna过多地存在于胞外体(灰色柱)中。在所有转化的b细胞类型中,mirna类型(mirbasev19)代表小rna库的约50%。然而在胞外体中,这一百分比仅为20%,表明mirna保留在细胞中。即使其他ncrna类型(即trna、pirna、rrna、yrna和穹窿体rna)的的分布在不同细胞类型间是有差异的(图1b),但衍生自该其他ncrna类型的rna元件通常富集于胞外体中。值得注意的是,trna和衍生自trna的片段在弥漫性大b细胞淋巴瘤(dlbcl)(mauteetal.,2013)和其他肿瘤组织(leeetal.,2009b)中表达失调。与lcl来源的胞外体相比,衍生自trna的片段在dlbcl来源的胞外体中高度富集(24%对7.4%;图1b)。lcl和淋巴瘤的胞外体之间最显著的区别是人类yrna片段的极端丰富(38%;图1)。为了研究观察到的小rna片段的读序丰度(图1)是否反映全长转录物的存在,我们对胞外体rna进行了半定量茎环rt-pcr,其中对胞外体rna进行测序。我们在胞外体中检测到高水平的完整的yrna种类(即rny1、rny3、rny4和rny5)和穹窿体rna1-1(图1)。在从人尿样分离的囊泡组分中,也检测到完整的yrna和穹窿rna(i.v.b.,d.k-l和d.m.p.,未发表的数据)。该数据支持了先前认为小细胞质调节rna可能有非细胞自主功能的意见(nolte-’thoenetal.,2012)。总体而言,我们发现了由rna聚合酶iii得到的yrna转录物及其片段具有胞外体掺入一致的倾向性,其对转化的b细胞而言似乎是最显著的。有趣的是,人类小rna,特别是yrna被包装于有包膜和无包膜的病毒颗粒中(garciaetal.,2009;khanetal.,2007;routhetal.,2012)。将小poliiirna从宿主细胞选择性招募到(逆转录)病毒颗粒和胞外体中,暗示共同的分子机制驱动了这一包装过程。这一认知可能有助于未来对控制胞外体生物发生和货物选择的细胞组分进行表征(koppers-lalicetal.,2012)。
高丰度和低丰度的microrna非随机地掺入胞外体中
虽然mirna的转录和生物发生是有据可查的(ebertandsharp,2012),但是仍然未完全了解控制mirna周转、亚细胞运输和经由胞外体的释放的机制,以及这些不同因素如何影响基因调节活性(ameresandzamore,2013)。
为了更好地了解mirna分布在细胞与胞外体之间可能的巨大差异,我们计算了所有细胞样品和配对胞外体(n=12)的标准化mirna读数之间的斯皮尔曼相关系数(spearmancorrelations),并确定了数据集之间的差异。通过聚类分析将lcl样品与淋巴瘤样品分离开。对于所有样品,斯皮尔曼相关系数(r)在0.48至0.81之间。lcl胞外体(r=0.76至0.8)和lcl细胞(r=0.72至0.76)显示出相对适度的样品变化性,表明其适合用作生物学重复。接下来,使用r程序包edger将定位的mirna计数拟合在广义线性模型中(robinsonetal.,2010),并比较细胞和胞外体中的各mirna的相对丰度。最丰富的细胞mirna(>10000读序/百万(rpm))通常代表胞外体中最丰富的mirna(表s2,数据1)。尽管在胞外体组分中的mirna分布明显受细胞mirna丰度的影响,但我们鉴定出了在细胞和胞外体之间分布不一致的一个mirna子集(错误发现率fdr=0.05)。
为了研究哪些mirna在lcl、伯基特淋巴瘤(burkittlymphoma,bl)和dlbcl背景中优先被保留或释放,将所有mirna根据胞外体相对于细胞的mirna读序(rpm)的富集倍数进行分组和排序。我们观察到分布最不一致的mirna优先被保留(15%至42%),而不是优先被释放(8%至15%)(图2a)。保留和释放的mirna在其细胞组分与胞外体组分中的重现证明,所有样品组所共有的释放的mirna的数目与从概率中预期的(z-分数=26.2)显著不同。重要的是,许多“释放的”mirna具有100~1000rpm以上的高相对丰度(图2b~图2c),其足以抑制基因调节活性(mullokandovetal.,2012)。
为了评估被定义为“保留的”或“释放的”的mirna的分布是否可通过其他方法检测,我们制备了新批次的b细胞分泌的胞外体(生物学重复),并从细胞和胞外体中提取了rna。接下来,我们进行了基于茎环的定量rt-pcr分析,并观察到释放的人mirnamir-451、mir-127-3p和mir-410在胞外体组分中高度富集,这与rna-seq方法的结果一致。引人注目的是,与优先“保留”的mirnamir-1275、mir-744和mir-130b(图2d)相比,mir-451和127-3p在细胞中几乎检测不到。mir-127-3p的排出可能与其作为b细胞淋巴瘤6(bcl6)mrna的负调节子的功能相关,所述b细胞淋巴瘤6(bcl6)mrna是淋巴瘤生成中的转录阻遏物(lujambioandesteller,2007;parekhetal.,2007)。事实上,mir-127-3p显示出肿瘤抑制活性(saitoetal.,2006),并且它的释放可以有利于b细胞淋巴瘤细胞的生长和存活。
人与病毒的mirna分布的差异
ebv-mirna为ebv感染的增殖性b细胞和ebv驱动的淋巴瘤提供了必要的生长优势(feederleetal.,2011;setoetal.,2010;vereideetal.,2013)。我们假设,与内源性肿瘤抑制因子mirna不同,ebv-mirna会被优先保留且不完全呈现在胞外体中。我们分组并分析了由转化的b细胞和淋巴瘤细胞中的ebv编码的44种mirna的分布(qiuetal.,2011)。与之前对ebv感染的b细胞系的rna-seq研究一致(rileyetal.,2012;skalskyetal.,2012),我们证实了ebv-mirna丰度变化很大,最高丰度的ebv-mirna属于bart-簇的mirna。在基于其分布频率进行读序标准化后,我们计算了细胞释放的所有mirna的量与保留在细胞中的量之间的比例(图4a)。而宿主mirna倾向于释放(17%是被细胞保留的),大多数病毒mirna不被分泌出去(54%是被强保留的)。这在图4b中示出,其中由ebv编码的病毒mirna表示为红色。这一观察结果与riley等人的结果一致,他们发现大量的ebvbart簇mirna靶向132个凋亡相关的细胞mrna(rileyetal.,2012),表明ebvmirna阻止细胞死亡。基因本体分析表明,已知抑制mapk/pi3k存活通路的许多宿主mirna被优先释放。所观察到的生理相关mirna的一致且非随机的分布与介导分选进入胞外体中的机制一致。
3'末端nta界定了体外和体内的mirna的保留或释放
来自mirna前体发夹结构的丰度最高的mirna序列通常用于定义相应的mirna,通常被称为成熟的mirna(mirbase)。然而,对各mirna读序的分析及其在相应mirna前体序列上的定位,表明在细胞和胞外体的rna组分中mirna的3'末端序列有很多变化。低效drosha或dicer介导的mirna前体的加工产生3'末端的长度变体(例如截短和延伸)。此外,mirna亚型通过核苷酸转移酶介导的转录后修饰(也称为非模板末端核苷酸添加(nta))主动产生(burroughsetal.,2010;morinetal.,2008;wymanetal.,2011)(图5a)。对mirna的3'末端变体的整体分析显示,3'末端长度的亚型在细胞和胞外体之间平均分布,而具有3'末端nta的mirna显示出差异分布。各nta类型(即a、u、c或g)与具有nta的所有读序的总和相比显示出腺苷酸化的mirna亚型在细胞组分中富集。相比之下,3'末端尿苷化的mirna在所分析的所有lcl样品的胞外体中过多出现(图5c)。为了分析nta修饰的读序(a,u,c或g)的比例在细胞和胞外体样品之间是否有显著改变,从所有12个样品中提取每一具有3'末端nta的mirna序列(lcln=6,bln=4,dlbcln=2),并拟合在回归模型(logisticmodel)中,其也针对细胞系效应进行校正(得到成对设计)。结果表明3'末端腺苷酸化的mirna亚型优先被保留(卡方检验,p<0.05),而所有其他的nta优先被释放(3'末端-u:卡方检验,p=0.02;3'末端-c:费舍尔(fisher)精确检验,p=0.04;和3'末端-g:fisher精确检验,p=0.02)。
除了nta分布整体分析,在三个lcl样品(即生物学上的一式三份)中,我们观察到3'末端腺苷酸化或尿苷化的mirna亚型在细胞和胞外体之间的差异是显著的(p=0.005,学生t检验)且未观察到成熟mirna(基于mirbase的注释)序列或未修饰的(延长的或截短的)亚型。为了分析3'末端nta对各mirna分布的影响,我们在lcl细胞中选择了100个大量表达的mirna,并与胞外体组分匹配进行成对分析。结果表明,具有3'末端腺苷酸化的各mirna序列与细胞组分更相关,而具有3'末端尿苷化的相同mirna更倾向于胞外体释放。因此我们得出结论,添加的核苷酸类型(即腺嘌呤或尿苷)影响mirna亚型在细胞和胞外体之间的分布。
对各mirna序列的更深入分析揭示了许多mirna的3'末端有不止一个的非模板核苷酸的延伸,其中各mirna序列允许在界定和注释的3'末端序列之外具有更多的错配和更长的侧翼区域(+6nt)。为了解决转录后添加的核苷酸的数量是否对分布具有累积效应,我们在考虑添加的核苷酸的数目的同时,首先计算了细胞和胞外体之间具有未修饰形式的特定nta类型的序列读数的倍数变化比例(被定义为排出系数(expelcoefficient);参见实验步骤)。3'末端腺苷酸化增加保留的概率,并且该概率随着添加的a的数目成比例地增加(图8b~图8c)。引人注目的是,三腺苷酸化的人mirna在lcl细胞中的频率比相应的胞外体中高5倍(p=0.03;非配对t检验),而三腺苷酸化的病毒mirna仅在lcl细胞组分中检测到(图8c)。3'末端腺苷酸化与病毒mirna在细胞中保留增加之间的关联,进一步证实了我们的观察,即病毒mirna显示出强的维持细胞相关的趋势(图4a~图4b)。重要的是,我们没有观察到广泛的3'末端的聚腺苷酸化(4个以上的腺嘌呤),其中3'末端的聚腺苷酸化(4个以上的腺嘌呤)是backes等人最近证明的mirna降解信号(backesetal.,2012)。
与3'末端腺苷酸化相比,在3'末端具有尿苷酸的小rna的不成比例的出现似乎定义了胞外体rna负荷。为了研究这一现象是否不限于培养的b细胞(图8f),我们使用胞外体分离方案从人尿液中收集并纯化了天然存在的细胞外囊泡(bijnsdorpetal.,2013;verweijetal.,2013)。我们对来自健康个体的六个尿液胞外体样品的小rna成分进行了测序(图10)。与在b细胞胞外体中观察到的3'末端尿苷化介导的mirna亚型分布完全相符(图8f;p值p<0.005;学生t检验),人尿液胞外体显著富含3'末端尿苷化的mirna(图8f;p值<0.0001;学生t检验)。总的来说,这些数据表明在体外以及在体内,细胞优先释放3'末端尿苷化的mirna,而mirna亚型的细胞保留取决于添加到mirna的3'末端的腺嘌呤核苷酸的数量。
3'末端nta对各mirna的胞外体分选的影响
我们的结果表明nta界定mirna在细胞和胞外体之间的分布。然而,nta对各mirna的影响可能不同。例如,大量胞外体富集的mirna486-5p、143-3p和101-3p的3'末端尿苷化读序示出高于胞外体组分中存在的所有尿苷化亚型的平均百分比(38%)。但同时,mir-486-5p似乎在成熟mirna水平分布相同(图7c;灰色)。值得注意的是,定位于成熟mir-486-5p序列的所有读序中的50%是腺苷酸化亚型或者是尿苷化亚型(图7c)。当使用lcl样品中所有mirna的加权平均值时,我们观察到,三尿苷化读序在lcl胞外体中的频率比细胞内容物高9倍(p=0.01;非配对t检验)。为了证实这种分布差异,我们开发了基于茎环的引物,其设计用于区分mir-486-5p的3'末端三腺苷酸化亚型和3'末端三尿苷化亚型,这些亚型比典型的(成熟的)基于mirbase的序列长三个核苷酸。如预期的,对3'末端未添加核苷酸的成熟mir-486-5p的检测显示在细胞和胞外体之间分布几乎相等,而3'末端三腺苷酸化亚型富集于细胞中(10倍)且3'末端三尿苷化亚型(5倍)富集于胞外体中(图7d)。这种独立的方案支持之前rnaseq的数据(图6),并且证明不同类型的3'末端转录后修饰可通过rt-pcr检测并反映细胞保留和胞外体分选过程。因此,大量的mirna及其亚型代表具有独特分布特性的亚群,其中的独特分布特性取决于3'末端转录后修饰的类型和程度。
除了人类和病毒的mirna,我们发现腺苷酸化和尿苷化也可以发生在源自小细胞质yrna的加工片段的3'末端(图9)。重要的是,大多数3'末端修饰的小rna分子在胞外体分选方面遵循与mirna亚型相同的命运,这表明作为分选信号的nta修饰不受mirna类别的限制。这些结果强调了3'末端nta的相关性,并且扩展了我们对于酶的核苷酸转移酶家族的小rna底物多样性的知识(martinandkeller,2007)。
细胞和胞外体的全部mirna之间的比较为大量mirna经由胞外体的选择性排出和释放提供了决定性证据。此外,我们观察到3'末端腺苷酸化的程度似乎阻碍mirna亚型经由胞外体的掺入和分泌。这一发现与这种特定修饰类型增加特定mirna的稳定性和活性的观点(burnsetal.,2011;d’ambrogioetal.,2012;jonesetal.,2009)一致。重要的是,我们发现了3'末端尿苷化的mirna亚型具有相反的行为。我们得出结论,mirna的运输、分选和经由胞外体的释放(这显而易见地也天然发生在人尿液囊泡中)部分通过nta的转录后修饰来界定。尽管这些细微的3'末端修饰在选择性mirna周转、丰度和活性中的确切作用还没有完全理解并且是复杂的(ameresandzamore,2013),但目前它们的功能意义正在被阐明(baccarinietal.,2011;d’ambrogioetal.,2012;jonesetal.,2009;katohetal.,2009;rüeggerandgroβhans,2012;scottandnorbury,2013)。我们的研究提供了对胞外体的小rna负荷选择的进一步理解,并提供了研究与胞外体生物学相关的mirna加工、修饰和周转的基础理论。
实验步骤
用于深度测序的rna样品的制备
对于每一细胞或胞外体的样品,根据制造商的说明书(illuminasandiegocausa)使用truseq小rna样品制备(truseqsmallrnasampleprep)制备等量的输入rna(600ngrna)用于测序。在agilent2100生物分析仪(agilent,santaclaracausa)上测量序列文库,并且每次运行时将多达12个样品等摩尔合并在一起。在hiseq2000(illuminasandiegocausa)运行配对末端100循环(pe100)以进行测序。
具有非模板核苷酸添加的rna读序的检测和统计分析
通过逐步分析检索mirna亚型或异构体。简言之,为了检测定位的rna元件的3'-末端的nta,我们从读序的第18位核苷酸开始,将读序序列与比对的mirna前体序列进行比较。如果算法在第18位后发现读序和微小rna前体之间的错配位置,则从错配位置到读序末端来进一步分析读序,同时所有后续核苷酸需要与错配位置处的那个核苷酸相同。如果遇到不同的核苷酸,则在下一个错配位置继续搜索或将读序标记为无nta。在检测了所有nta读序之后,我们通过将以给定核碱基(a,u,c和g)的nta结尾的读序的数值除以定位至mirna的读序的总数值,计算每个样品的加权平均值。每个mirna和每个样品,除了定位至该mirna的读序的总数值外,我们计算以给定核碱基的nta结尾的读序的总数值。因此,对于每个核碱基,我们可以使用逻辑回归模型中的相对于总数值的读序数值,其中逻辑回归模型还包括样本类型(细胞或胞外体)以及细胞系(提供配对设计)。基于这个模型,我们取得了细胞和胞外体之间,具有给定核苷酸的nta的mirna计数的比例间的差异的p值。在拟合每个mirna的此模型后,将本-霍氏(benjamini-hochberg)的伪发现率(fdr)应用于p值以校正多重测试。
具有非模板核苷酸添加的小rna的排出趋势
采用以下方式,对转录后修饰的读序(nta)在细胞和胞外体之间的分布进行评估。为了测试mirna相关的读序是否具有掺入到胞外体中或保留在细胞内的趋势,我们将其与给定mirna(被定义为除了属于所分析的mirna亚型外的所有其他读序)的基线分布趋势进行了比较。我们通过计算以特定类型的非模板核苷酸(被定义为nta)结尾的读序和那些没有末端修饰的(被定义为无nta)结尾的读序在胞外体和细胞之间的倍数变化的比例,来定义排出系数(ec)。小ncrna序列的nta读序和无nta读序具有相同的释放或保留的趋势时,系数将趋近于0。如果nta读序与无nta读序相比具有更强的被释放的趋势,则为正值。最后,负系数表明nta读序与无nta读序相比具有更强的保留在细胞内的趋势。
胞外体的收集步骤
为了收集胞外体,将每一细胞系在补充有10%去除胞外体的fbs的rpmi-1640中进行连续三轮培养,随后收获含胞外体的上清液(一轮收集表示一个含有胞外体的制备物;200ml培养基中有100×106细胞)。通过台盼蓝排除法对细胞死亡进行常规分析,生存力≤90%的培养物不考虑用于胞外体收集。使用如(verweijetal.,2013)描述的差速离心方法,从上清液中分离和纯化胞外体。简言之,最后一步包括以70000×g离心1小时使胞外体沉淀成块,并在进行最终的超速离心步骤(70000×g)之前用pbs清洗沉淀块。将成块的外胞体溶解于100μl的pbs中并储存在-80℃。如前所述((pegteletal.,2010;verweijetal.,2011),通过免疫印迹分析所有胞外体制备物以确认胞外体的存在和纯度(数据未显示)。为了提取尿液的胞外体,在签署知情同意后从健康男性(n=6)收集新鲜尿液样品(50ml体积)。这些健康男性是在泌尿科参与与该项目无关的研究的患者对照组(vu大学医学中心,阿姆斯特丹,荷兰)。通过差速离心分离胞外体。首先,将尿液以500×g离心20分钟,以2000×g离心20分钟,10000×g离心30分钟,100000×g离心1.5小时,再用pbs洗涤一次。将完整的胞外体收集在pbs中,用rna酶(rnase)a(400ng/ml,sigma-aldrichchemicals,兹韦恩德雷赫特,荷兰)处理,然后进行rna分离。
rna提取、质量评估和半定量pcr测定
从细胞和从配对的胞外体沉淀块中提取rna。用400ng/ulrnasea(sigma)在37℃预处理胞外体制备物1小时。通过trizol试剂(lifetechnologies),从所有的胞外体制备物提取总rna。当预期产量低时,在异丙醇沉淀步骤之前加入5μl糖原(roche)。为了能比较细胞样品和配对的胞外体(已知包含多数大小<200nt的小rna变体)之间的小rna谱,使用基于玻璃纤维滤器的方法(mirvanamirna分离试剂盒;lifetechnologies)提取细胞的rna,以富集存在于细胞样品中的小rna种类。提取后,所有细胞rna样品都包括小rna种类(<200nt)。小细胞和胞外体rna谱显示其大小分布的模式相似。使用小rna芯片平台(agilent2100生物分析仪)测定所提取的小rna(细胞和胞外体)的质量和数量。
使用sybr绿色通过基于茎-环的半定量逆转录酶pcr(rt-pcr)检测细胞样品和胞外体样品中的全长小非编码rna的存在,并通过lightcycler480(roche)进行分析。该方法使用用于rt反应的茎-环引物。设计茎-环引物以与感兴趣的小rna的3'-末端最后8个核苷酸的一段互补,且使用时终浓度为12.5nm。使用taqmanmicrornart试剂盒(appliedbiosystems),并按照制造商的说明书孵育反应物。通过rna特异性正向引物,并且通过针对靶标的重叠区特异的反向引物和茎-环rt引物扩增产物。扩增产物通过lightcycler480软件(roche)进行分析。按照制造商的说明书,通过taqmanmicrorna测定和customtaqman小rna测定(lifetechnologies)检测细胞和胞外体的mirna。从经appliedbiosystems7500fastreal-timepcr系统及所提供的分析软件所分析的样品中,获得所有mirna的数据。所有rt-pcr反应都使用等量的rna。除了图2d中使用δδct方法将ct值对于mir-92a进行标准化外,结果以图4b中的重复实验的两个技术重复的平均循环阈值(ct)值(平均ct值)来表示。ct值大于40的样品视为阴性。
深度测序数据和小rna的表达谱的分析
使用基于miranalyzer程序(hackenbergetal.,2011)(包括额外的分析步骤和自定义的脚本)的方式分析小rna的表达谱。简言之,对fastq格式的读序的预处理包括:i)接头剪切(需要检测到至少10nt的接头,允许读序和接头序列之间有1个错配),ii)删除短于15nt的清除后的读序,和iii)将具有相同序列的所有读序缩小成一个条目(唯一读序)。唯一读序的读序计数是表示相应rna分子已经被测序了多少次的数目。
在接头剪切及唯一读序分组后,利用鲍蒂(bowtie)算法(langmeadetal.,2009)将读序与人(grch37补丁发布5;下载自ucsc上的hg19)和ebv基因组(基因库登录号nc_007605)的合并索引(pooledindex)进行比对。我们使用长度为19bp的比对种子,允许1个错配,仅取回那些定位至基因组中的小于40个位置的比对结果。为了从那些满足标准的比对结果中仅取回最佳比对结果,我们使用了如hackenberg等人(hackenbergetal.,2011)所解释的种子扩展方法。需要注意的是,bowtie种子比对方法仅允许比对读序的前l个碱基。常常通过在小rna的3'-末端的rna测序,来检测腺嘌呤(a)、尿嘧啶(u)、胞嘧啶(c)和鸟嘌呤(g)的非基因组模板的核苷酸添加(nta)。转录后添加的碱基不存在于基因组序列中,因此比对时将检测为错配。种子比对能检测到在种子区域外作为错配的那些读序,且不会解释为允许的错配。在与基因组(人和ebv基因组组合)比对后,几个注释用于小rna品种的表达谱分析。通常,读序位置必须完全位于由小rna注释所界定的区域内:[染色体起始-3nt:染色体末端+6nt]。为了能够检测长度变体和nta,我们允许有侧翼区域。给定小rna的表达值只是定位在相应基因组区域内的所有读序的总和。此外,我们通过将各读序数(rc)除以定位的染色体位置的数目,来计算调整后的rc。最后,我们计算对于各小rna元件的每百万表达值的读序(rpm),利用定位至给定rna种类的读序总进行标准化。rpm是使表达值不依赖于读序总数的最常见的标准化类型。
为了对定位至人和ebv基因组的rna元件提供注释,针对当前已知的数据库分析了所有定位的读序:1)来自mirbase第19版的成熟mirna序列和mirna前体序列(kozomaraandgriffiths-jones,2011),2)2013年2月6日下载的ncbi参考序列人类refseq基因(pruittetal.,2012),3)来自基因组trna数据库的trna序列(chanandlowe,2009),4)通过从ucsc下载的repeatmasker算法检测的重复衍生序列,和5)从ncbi核苷酸数据库下载piwirna(pirna)。对于fasta格式中的那些注释,我们通过将序列定位至基因组而得到基因组坐标。
比较细胞样本和胞外体样本的统计分析
rna-seq文库每一细胞系涉及一对文库,对应于来自全细胞的rna或仅来自胞外体的rna。为了比较各mirna在细胞样品和胞外体样品之间的丰度,使用r程序包edger(robinsonetal.,2010)将观察到的计数拟合在广义线性模型中,其包括用于配对设计的细胞系,以及样品类型(细胞或胞外体)。该模型不仅包括常见和趋势离散度,而且还包括tagwise离散度评估,从而允许由于文库间波动而产生额外的可变性。使用benjamini-hochberg的伪发现率(benjamini,2010),对多次检验的p值进行校正,其中p值对应于样本类型效应的似然比检验统计量。此外,我们还将观察到的6个样本之间的交集与随机期望进行比较。简言之,我们i)计算表达值在所有6个样品的胞外体和细胞之间的log2比例,ii)提取所有排出的(log2>=2)mirna,iii)提取给定组的共有mirna,即,在该组的所有样品中都被排出的那些mirna,iv)每一条件下随机提取10000次xmirna(x是观察到的数目,即排出的mirna),v)我们检测10000次随机运行中每一次的共有mirna的数目,vi)从10000次随机运行中,我们计算随机共有的rna的平均值和标准偏差,其作为期望值及其在随机假设下的波动,vii)利用观察到的共有rna的数量和随机平均值和标准偏差,我们计算z分数来评估统计学意义。我们认为z分数为1.645或更高,对应于p≤0.05,具有显著意义。
实施例2
我们选择了明确的细胞肾细胞癌(ccrcc)的实验,由11个健康(非肿瘤肾皮质-nrc)和22个明确的细胞肾细胞癌样品组成(osanto,s.etal.plosone2012)。此外,22位ccrcc患者属于3个预后亚组,即在诊断时没疾病未复发、有复发以及具有转移性疾病(iv期)。从shortreadarchive(sra)下载sra登录号为srp012546的数据。为了描述mirna表达模式并确定isomir的程度,我们扩展了我们以前开发的广泛使用的程序miranalyzer,其注释了复杂(小)rnaseq数据(hackenberg,m.etal.nucleicacidres.2009&2011)。在接头剪切后,将唯一读序定位至人类基因组。为了检测所有序列变体,我们允许与典型的序列相比有波动:在5'末端的3nt波动和在3'末端的5nt波动。
在检测到属于给定mirna的所有读序后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta)和iii)源自典型序列的5'和/或3'序列长度变体(例如截短或延伸)。计算各mirna和isomirna类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。
在用我们的方式分析数据之后,我们比较了所有4个实验组(健康、未复发、复发、iv期)之间观察到的isomir比例。为了获得那些在两组之间存在统计学显著差异的mirna,我们采用了标准t检验。我们发现临床相关组之间存在腺嘌呤添加(腺苷酸化)的明显差异。我们提取了4个mirna,其在至少一个组(>=35%)中和在至少一个显著比较中显示出非常高的腺苷酸化百分比的读序。这4个成熟的mirna是:mir-199b-3p,mir-125b-5p,mir-210-3p和mir-151a-3p。图11描述了所有4个不同患者组的这四种mirna的isomirna的比例。
初步结果的简要概述
1.不同的mirna显示腺苷酸化频率和表达水平(包括对应于成熟mirna序列的稳定的mir-210-3p)之间有强相关性(d’ambrogioetal.cellreports2012)。在rcc进展期间mir-210-3p水平增加,这可能是通过转录后修饰以3'-末端腺苷酸化的形式使mirna稳定化的结果。
2.在iv期和健康个体之间,成熟的mir-199b-3p未差异化表达(参见图11),然而在腺苷酸化模式中存在高显著差异(p值:3.83e-06)。此外,mir-199b-3p靶向4种与肾癌发生高度相关的基因(med6、krt7、met、junb)。因此推定该mirna参与肿瘤发生,但是通过传统分析途径不会检测到该mirna,强调了独立地确定isomir含量的重要性。
3.患ccrrc的经治疗未复发的个体显示出更接近健康个体的mir-199b-3p腺苷酸化比例(35%,p值=0.13),而复发的个体的腺苷酸化比例确实接近iv期的个体(22%,p值=4.72*10-4)。对于mir-125b-5p,可观察到非常相似的模式(参见图11)。这表明isomir具有意想不到的预后生物标记物的潜能。
实施例3:生物流体中的isomir区分癌症亚型和健康患者
尿液收集自4名健康的志愿者和9名诊断为前列腺癌的患者。如实施例1所述收集细胞外体并提取rna。如实施例1和实施例2那样,进行小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)5'和/或3'剪切变体。计算各mirna和isomir类型的isomir比例。图12中描绘了最有统计学意义的比值(由p值确定)。变体与典型mirna序列的比例在健康患者和那些前列腺癌患者之间是显著的。
实施例4:用于从人血浆中一步分离囊泡的尺寸排阻色谱(sec)
测量血液中的循环ncrna(例如mirna)可用于早期癌症检测、预后和监测。对组织和培养细胞进行的全面rna测序揭示了复杂且动态的ncrna的全部组成。已经鉴定了超过2000个成熟mirna种类,其中许多已经在循环和其他生物流体(例如唾液和尿液)中检测到。然而,一个基因组位点可以产生许多功能上不同的ncrna变体。
血液中的大多数细胞外ncrna与可溶性生物化学组分相关,其中可溶性生物化学组分包括蛋白质复合物、脂质囊泡(ldl和hdl)和细胞外囊泡。由于技术限制,在总血清或血浆中进行全面的ncrna表达谱分析是冗长的。制备循环的小rna及其变体的小rna文库需要可用于接头连接的完整ncrna种类具有足够高的富集和纯度。
为了增加测量与个体健康状况相关的ncrna的信噪比,我们采用如下方式测量经纯化的胞外体囊泡(ev)和蛋白质组分中的mirna。
琼脂糖cl-2b的预处理
·在烧杯中倒入20ml琼脂糖cl-2b(每柱),让琼脂糖cl-2b静置至少15分钟。
·弃去上清液
·加入15ml缓冲液,涡旋混匀
·使琼脂糖静置至少15分钟
·重复清洗两次
·弃去上清液,加入10ml缓冲液
柱的准备
·使用10ml注射器
·将尼龙软管(pantyhose)压入注射器的出口中
·用移液管将洗过的琼脂糖移入柱中(使用塑料的巴斯德(pasteur)移液管)
·使琼脂糖静置,避免柱子变干;柱子应该精确地垂直,没有角度(使用水平仪)
·加入琼脂糖直至达到10ml堆积的琼脂糖
·避免柱子变干。添加缓冲液,直至使用或盖上注射器插头(几个小时内使用柱子)
样品上样
让缓冲液几乎完全流入琼脂糖柱(但防止柱子变干)
-在柱子上上样1.5ml血浆
-立即开始收集0.5ml样品
-当血浆样品几乎完全进入琼脂糖时,小心地加入缓冲液直到完全充满注射器
-重复添加缓冲液,直到收集到所有组分(多达26种组分)
测序来自临床样品的循环小rna
-使用trizol分离方法从单一组分中分离总rna
-在生物分析仪上测量rna并使用rt-pcr进行质量控制
-按照制造商的指南(illuminatruseq)制备小rna文库
-按照制造商的说明书(illuminahiseq)制备序列文库
对获自典型霍奇金淋巴瘤患者的血浆进行如上所述的sec。如图14所示,sec能够从血浆中一步分离循环的ev。mirna的分布在ev组分和蛋白质/hdl组分间是不同的(图14e)。
使用上述方法,我们发现了多个囊泡相关的mirna以及蛋白质组分相关的mirna,其能监测霍奇金患者的治疗响应。这些结果在实施例5中进行了讨论。
实施例5:从霍奇金淋巴瘤的血浆中分离的ev
如实施例4所述,对获自霍奇金淋巴瘤患者的血浆进行sec。结果如图16~图18所示。
实施例6:生物流体中的isomir将健康个体与霍奇金淋巴瘤患者区分开。如实施例4和实施例5中所述,进行了细胞外囊泡和蛋白质结合rna组分的分离。如实施例1和实施例2中所述,进行了小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)源自典型序列的5'和/或3'序列变体(例如截短或延伸)。计算各mirna和isomir类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。图15中描绘了最有统计学意义的比值(由p值确定)。变体与典型序列的比例在健康个体和那些患有霍奇金淋巴瘤的个体之间是显著的。
实施例7:生物流体中的isomir将乳腺癌ii期与疾病iii期区分开,并且将疾病复发的乳腺癌患者与无复发的乳腺癌患者区分开。
数据源和实验程序是公开可用的,并可从http://www.ncbi.nlm.nih.gov/sra?term=srp027589和http://www.ncbi.nlm.nih.gov/bioproject?linkname=sra_bioproject&from_uid=455584获得。
分析血清中公开可用的ncrna序列数据,以确定ncrna变体的比例是否可用于分类健康状况。如实施例2进行小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)源自典型序列的5'和/或3'序列变体(例如截短或延伸)。计算各mirna和isomir类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。表6描绘了最具统计学意义的比值(由p值确定)。变体与典型序列的比例在ii期患者与iii期乳腺癌患者之间是显著的。
实施例8:组织中的isomir将癌旁的非肿瘤组织与睾丸生殖细胞肿瘤区分开
数据源和实验程序是公开可用的,并可从http://www.ncbi.nlm.nih.gov/sra?term=srp007946和http://www.ncbi.nlm.nih.gov/bioproject/154993获得。分析血清中公开可用的ncrna序列数据,以确定ncrna变体的比例是否可用于分类健康状况。
如实施例2进行小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)源自典型序列的5'和/或3'序列变体(例如截短或延伸)。计算各mirna和isomir类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。表7描绘了最具统计学意义的比值(由p值确定)。变体与典型序列的比例在癌旁组织和来自睾丸生殖细胞肿瘤的组织之间是显著的。
实施例9:组织中的isomir将结直肠正常组织与结直肠肿瘤组织和结直肠癌相关转移组织区分开
数据源和实验程序是公开可用的,并可从http://www.ncbi.nlm.nih.gov/traces/sra/?study=srp022054和http://www.ncbi.nlm.nih.gov/bioproject/prjna201245和
如实施例2进行小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)源自典型序列的5'和/或3'序列变体(例如截短或延伸)。计算各mirna和isomir类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。表8描绘了最具统计学意义的比值(由p值确定)。变体与典型序列的比例在正常结直肠组织和来自结直肠肿瘤的组织之间,在正常结直肠组织和结直肠肿瘤相关转移之间,以及在结直肠肿瘤组织和结直肠肿瘤相关转移之间都是显著的。
实施例10:生物流体中的isomir将健康个体与诊断为阿尔茨海默病的患者区分开
数据源和实验程序是公开可用的,并可从http://www.ncbi.nlm.nih.gov/sra?term=srp022043和http://www.ncbi.nlm.nih.gov/bioproject?linkname=sra_bioproject&from_uid=87967和leidingerpetal.,"abloodbased12-mirnasignatureofalzheimerdiseasepatients.",genomebiol,2013jul29;14(7):r78中获得。
分析血清中公开可用的ncrna序列数据,以确定ncrna变体的比例是否可用于分类健康状况。如实施例2进行小ncrna的表达谱分析和数据分析。
在检测到属于给定mirna的所有读序之后,对读序进行分级分类(按isomir类型分配):i)典型的mirna序列,ii)非模板添加(nta),和iii)源自典型序列的5'和/或3'序列变体(例如截短或延伸)。计算各mirna和isomir类型的isomir比例。我们将它们定义为属于给定isomir类型的读序数除以定位至给定mirna的读序总数(典型读序的计数加上所有isomir)。表9描绘了最具统计学意义的比值(由p值确定)。变体与典型序列的比例在健康个体与那些阿尔茨海默病患者之间是显著的。
参考文献
amaral,p.p.,dinger,m.e.,mercer,t.r.,andmattick,j.s.(2008).theeukaryoticgenomeasanrnamachine.science319,1787–1789.
ameres,s.l.,andzamore,p.d.(2013).diversifyingmicrornasequenceandfunction.nat.rev.mol.cellbiol.14,475–488.
ameres,s.l.,martinez,j.,andschroeder,r.(2007).molecularbasisfortargetrnarecognitionandcleavagebyhumanrisc.cell130,101–112.
ameres,s.l.,horwich,m.d.,hung,j.-h.,xu,j.,ghildiyal,m.,weng,z.,andzamore,p.d.(2010).targetrna-directedtrimmingandtailingofsmallsilencingrnas.science328,1534–1539.
baccarini,a.,chauhan,h.,gardner,t.j.,jayaprakash,a.d.,sachidanandam,r.,andbrown,b.d.(2011).kineticanalysisrevealsthefateofamicrornafollowingtargetregulationinmammaliancells.curr.biol.21,369–376.
backes,s.,shapiro,j.s.,sabin,l.r.,pham,a.m.,reyes,i.,moss,b.,cherry,s.,andtenoever,b.r.(2012).degradationofhostmicrornasbypoxviruspoly(a)polymeraserevealsterminalrnamethylationasaprotectiveantiviralmechanism.cellhostmicrobe12,200–210.
balaj,l.,lessard,r.,dai,l.,cho,y.-j.,pomeroy,s.l.,breakefield,x.o.,andskog,j.(2011).tumourmicrovesiclescontainretrotransposonelementsandamplifiedoncogenesequences.nat.commun.2,180.
vanbalkom,b.w.m.,dejong,o.g.,smits,m.,brummelman,j.,denouden,k.,debree,p.m.,vaneijndhoven,m.a.j.,pegtel,d.m.,stoorvogel,w.,würdinger,t.,etal.(2013).endothelialcellsrequiremir-214tosecreteexosomesthatsuppresssenescenceandinduceangiogenesisinhumanandmouseendothelialcells.blood121,3997–4006,s1–15.
bellingham,s.a.,coleman,b.m.,andhill,a.f.(2012).smallrnadeepsequencingrevealsadistinctmirnasignaturereleasedinexosomesfromprion-infectedneuronalcells.nucleicacidsres.40,10937–10949.
bijnsdorp,i.v,geldof,a.a.,lavaei,m.,piersma,s.r.,vanmoorselaar,r.j.a.,andjimenez,c.r.(2013).exosomalitga3interfereswithnon-cancerousprostatecellfunctionsandisincreasedinurineexosomesofmetastaticprostatecancerpatients.j.extracell.vesicles2.
burns,d.m.,d’ambrogio,a.,nottrott,s.,andrichter,j.d.(2011).cpebandtwopoly(a)polymerasescontrolmir-122stabilityandp53mrnatranslation.nature473,105–108.
burroughs,a.m.,ando,y.,dehoon,m.j.l.,tomaru,y.,nishibu,t.,ukekawa,r.,funakoshi,t.,kurokawa,t.,suzuki,h.,hayashizaki,y.,etal.(2010).acomprehensivesurveyof3’animalmirnamodificationeventsandapossiblerolefor3'adenylationinmodulatingmirnatargetingeffectiveness.genomeres.20,1398–1410.
chairoungdua,a.,smith,d.l.,pochard,p.,hull,m.,andcaplan,m.j.(2010).exosomereleaseofβ-catenin:anovelmechanismthatantagonizeswntsignaling.j.cellbiol.190,1079–1091.
chitwood,d.h.,andtimmermans,m.c.p.(2010).smallrnasareonthemove.nature467,415–419.
chuma,s.,andpillai,r.s.(2009).retrotransposonsilencingbypirnas:ping-pongplayersmarktheirsub-cellularboundaries.plosgenet.5,e1000770.
cullen,b.r.(2009).viralandcellularmessengerrnatargetsofviralmicrornas.nature457,421–425.
d’ambrogio,a.,gu,w.,udagawa,t.,mello,c.c.,andrichter,j.d.(2012).specificmirnastabilizationbygld2-catalyzedmonoadenylation.cellrep.2,1537–1545.
ebert,m.s.,andsharp,p.a.(2012).rolesformicrornasinconferringrobustnesstobiologicalprocesses.cell149,515–524.
feederle,r.,haar,j.,bernhardt,k.,linnstaedt,s.d.,bannert,h.,lips,h.,cullen,b.r.,anddelecluse,h.-j.(2011).themembersofanepstein-barrvirusmicrornaclustercooperatetotransformblymphocytes.j.virol.85,9801–9810.
fernandez-valverde,s.l.,taft,r.j.,andmattick,j.s.(2010).dynamicisomirregulationindrosophiladevelopment.rna16,1881–1888.
garcia,e.l.,onafuwa-nuga,a.,sim,s.,king,s.r.,wolin,s.l.,andtelesnitsky,a.(2009).packagingofhostmyrnasbymurineleukemiavirusmayoccurearlyinyrnabiogenesis.j.virol.83,12526–12534.
gibbings,d.j.,ciaudo,c.,erhardt,m.,andvoinnet,o.(2009).multivesicularbodiesassociatewithcomponentsofmirnaeffectorcomplexesandmodulatemirnaactivity.nat.cellbiol.11,1143–1149.
guasparri,i.,bubman,d.,andcesarman,e.(2008).ebvlmp2aaffectslmp1-mediatednf-kappabsignalingandsurvivaloflymphomacellsbyregulatingtraf2expression.blood111,3813–3820.
guduric-fuchs,j.,o’connor,a.,camp,b.,o’neill,c.l.,medina,r.j.,andsimpson,d.a.(2012).selectiveextracellularvesicle-mediatedexportofanoverlappingsetofmicrornasfrommultiplecelltypes.bmcgenomics13,357.
guo,l.,yang,q.,lu,j.,li,h.,ge,q.,gu,w.,bai,y.,andlu,z.(2011).acomprehensivesurveyofmirnarepertoireand3’additioneventsintheplacentasofpatientswithpre-eclampsiafromhigh-throughputsequencing.plosone6,e21072.
jones,m.r.,quinton,l.j.,blahna,m.t.,neilson,j.r.,fu,s.,ivanov,a.r.,wolf,d.a.,andmizgerd,j.p.(2009).zcchc11-dependenturidylationofmicrornadirectscytokineexpression.nat.cellbiol.11,1157–1163.
kai,z.s.,andpasquinelli,a.e.(2010).micrornaassassins:factorsthatregulatethedisappearanceofmirnas.nat.struct.mol.biol.17,5–10.
katoh,t.,sakaguchi,y.,miyauchi,k.,suzuki,t.,kashiwabara,s.-i.,baba,t.,andsuzuki,t.(2009).selectivestabilizationofmammalianmicrornasby3’adenylationmediatedbythecytoplasmicpoly(a)polymerasegld-2.genesdev.23,433–438.
khan,m.a.,goila-gaur,r.,opi,s.,miyagi,e.,takeuchi,h.,kao,s.,andstrebel,k.(2007).analysisofthecontributionofcellularandviralrnatothepackagingofapobec3gintohiv-1virions.retrovirology4,48.
koppers-lalic,d.,hogenboom,m.m.,middeldorp,j.m.,andpegtel,d.m.(2012).virus-modifiedexosomesfortargetedrnadelivery;anewapproachinnanomedicine.adv.drugdeliv.rev.
kosaka,n.,iguchi,h.,yoshioka,y.,takeshita,f.,matsuki,y.,andochiya,t.(2010).secretorymechanismsandintercellulartransferofmicrornasinlivingcells.j.biol.chem.285,17442–17452.
kosaka,n.,iguchi,h.,hagiwara,k.,yoshioka,y.,takeshita,f.,andochiya,t.(2013).neutralsphingomyelinase2(nsmase2)-dependentexosomaltransferofangiogenicmicrornasregulatecancercellmetastasis.j.biol.chem.288,10849–10859.
lee,y.s.,pressman,s.,andress,a.p.,kim,k.,white,j.l.,cassidy,j.j.,li,x.,lubell,k.,lim,d.h.,cho,i.s.,etal.(2009a).silencingbysmallrnasislinkedtoendosomaltrafficking.nat.cellbiol.11,1150–1156.
lee,y.s.,shibata,y.,malhotra,a.,anddutta,a.(2009b).anovelclassofsmallrnas:trna-derivedrnafragments(trfs).genesdev.23,2639–2649.
lujambio,a.,andesteller,m.(2007).cpgislandhypermethylationoftumorsuppressormicrornasinhumancancer.cellcycle6,1455–1459.
martens-uzunova,e.s.,olvedy,m.,andjenster,g.(2013).beyondmicrorna--novelrnasderivedfromsmallnon-codingrnaandtheirimplicationincancer.cancerlett.340,201–211.
martin,g.,andkeller,w.(2007).rna-specificribonucleotidyltransferases.rna13,1834–1849.
maute,r.l.,schneider,c.,sumazin,p.,holmes,a.,califano,a.,basso,k.,anddalla-favera,r.(2013).trna-derivedmicrornamodulatesproliferationandthednadamageresponseandisdown-regulatedinbcelllymphoma.proc.natl.acad.sci.u.s.a.110,1404–1409.
mittelbrunn,m.,andsánchez-madrid,f.(2012).intercellularcommunication:diversestructuresforexchangeofgeneticinformation.nat.rev.mol.cellbiol.13,328–335.
mittelbrunn,m.,gutiérrez-vázquez,c.,villarroya-beltri,c.,gonzález,s.,sánchez-cabo,f.,gonzález,
montecalvo,a.,larregina,a.t.,shufesky,w.j.,stolz,d.b.,sullivan,m.l.g.,karlsson,j.m.,baty,c.j.,gibson,g.a.,erdos,g.,wang,z.,etal.(2012).mechanismoftransferoffunctionalmicrornasbetweenmousedendriticcellsviaexosomes.blood119,756–766.
morin,r.d.,o’connor,m.d.,griffith,m.,kuchenbauer,f.,delaney,a.,prabhu,a.-l.,zhao,y.,mcdonald,h.,zeng,t.,hirst,m.,etal.(2008).applicationofmassivelyparallelsequencingtomicrornaprofilinganddiscoveryinhumanembryonicstemcells.genomeres.18,610–621.
mullokandov,g.,baccarini,a.,ruzo,a.,jayaprakash,a.d.,tung,n.,israelow,b.,evans,m.j.,sachidanandam,r.,andbrown,b.d.(2012).high-throughputassessmentofmicrornaactivityandfunctionusingmicrornasensoranddecoylibraries.nat.methods9,840–846.
nolte-’thoen,e.n.m.,buermans,h.p.j.,waasdorp,m.,stoorvogel,w.,wauben,m.h.m.,and’thoen,p.a.c.(2012).deepsequencingofrnafromimmunecell-derivedvesiclesuncoverstheselectiveincorporationofsmallnon-codingrnabiotypeswithpotentialregulatoryfunctions.nucleicacidsres.40,9272–9285.
palma,j.,yaddanapudi,s.c.,pigati,l.,havens,m.a.,jeong,s.,weiner,g.a.,weimer,k.m.e.,stern,b.,hastings,m.l.,andduelli,d.m.(2012).micrornasareexportedfrommalignantcellsincustomizedparticles.nucleicacidsres.40,9125–9138.
parekh,s.,polo,j.m.,shaknovich,r.,juszczynski,p.,lev,p.,ranuncolo,s.m.,yin,y.,klein,u.,cattoretti,g.,dallafavera,r.,etal.(2007).bcl6programslymphomacellsforsurvivalanddifferentiationthroughdistinctbiochemicalmechanisms.blood110,2067–2074.
pegtel,d.m.,cosmopoulos,k.,thorley-lawson,d.a.,vaneijndhoven,m.a.j.,hopmans,e.s.,lindenberg,j.l.,degruijl,t.d.,würdinger,t.,andmiddeldorp,j.m.(2010).functionaldeliveryofviralmirnasviaexosomes.proc.natl.acad.sci.u.s.a.107,6328–6333.
pegtel,d.m.,vandegarde,m.d.b.,andmiddeldorp,j.m.(2011).viralmirnasexploitingtheendosomal-exosomalpathwayforintercellularcross-talkandimmuneevasion.biochim.biophys.acta1809,715–721.
polikepahad,s.,andcorry,d.b.(2013).profilingofthelpercell-derivedsmallrnasrevealsuniqueantisensetranscriptsanddifferentialassociationofmirnaswithargonauteproteins1and2.nucleicacidsres.41,1164–1177.
qiu,j.,cosmopoulos,k.,pegtel,m.,hopmans,e.,murray,p.,middeldorp,j.,shapiro,m.,andthorley-lawson,d.a.(2011).anovelpersistenceassociatedebvmirnaexpressionprofileisdisruptedinneoplasia.plospathog.7,e1002193.
riley,k.j.,rabinowitz,g.s.,yario,t.a.,luna,j.m.,darnell,r.b.,andsteitz,j.a.(2012).ebvandhumanmicrornasco-targetoncogenicandapoptoticviralandhumangenesduringlatency.emboj.31,2207–2221.
robinson,m.d.,mccarthy,d.j.,andsmyth,g.k.(2010).edger:abioconductorpackagefordifferentialexpressionanalysisofdigitalgeneexpressiondata.bioinformatics26,139–140.
routh,a.,domitrovic,t.,andjohnson,j.e.(2012).hostrnas,includingtransposons,areencapsidatedbyaeukaryoticsingle-strandedrnavirus.proc.natl.acad.sci.u.s.a.109,1907–1912.
rüegger,s.,andgroβhans,h.(2012).micrornaturnover:when,how,andwhy.trendsbiochem.sci.37,436–446.
saito,y.,liang,g.,egger,g.,friedman,j.m.,chuang,j.c.,coetzee,g.a.,andjones,p.a.(2006).specificactivationofmicrorna-127withdownregulationoftheproto-oncogenebcl6bychromatin-modifyingdrugsinhumancancercells.cancercell9,435–443.
scott,d.d.,andnorbury,c.j.rnadecayvia3’uridylation.biochim.biophys.acta1829,654–665.
scott,d.d.,andnorbury,c.j.(2013).rnadecayvia3′uridylation.biochim.biophys.acta-generegul.mech.1829,654–665.
seto,e.,moosmann,a.,
sim,s.,weinberg,d.e.,fuchs,g.,choi,k.,chung,j.,andwolin,s.l.(2009).thesubcellulardistributionofanrnaqualitycontrolprotein,theroautoantigen,isregulatedbynoncodingyrnabinding.mol.biol.cell20,1555–1564.
skalsky,r.l.,corcoran,d.l.,gottwein,e.,frank,c.l.,kang,d.,hafner,m.,nusbaum,j.d.,feederle,r.,delecluse,h.-j.,luftig,m.a.,etal.(2012).theviralandcellularmicrornatargetomeinlymphoblastoidcelllines.plospathog.8,e1002484.
song,l.,lin,c.,gong,h.,wang,c.,liu,l.,wu,j.,tao,s.,hu,b.,cheng,s.-y.,li,m.,etal.(2013).mir-486sustainsnf-κbactivitybydisruptingmultiplenf-κb-negativefeedbackloops.cellres.23,274–289.
umezu,t.,ohyashiki,k.,kuroda,m.,andohyashiki,j.h.(2013).leukemiacelltoendothelialcellcommunicationviaexosomalmirnas.oncogene32,2747–2755.
vereide,d.t.,seto,e.,chiu,y.-f.,hayes,m.,tagawa,t.,grundhoff,a.,hammerschmidt,w.,andsugden,b.(2013).epstein-barrvirusmaintainslymphomasviaitsmirnas.oncogene.
verweij,f.j.,vaneijndhoven,m.a.j.,middeldorp,j.,andpegtel,d.m.(2013).analysisofviralmicrornaexchangeviaexosomesinvitroandinvivo.methodsmol.biol.1024,53–68.
villarroya-beltri,c.,gutiérrez-vázquez,c.,sánchez-cabo,f.,pérez-hernández,d.,vázquez,j.,martin-cofreces,n.,martinez-herrera,d.j.,pascual-montano,a.,mittelbrunn,m.,andsánchez-madrid,f.(2013).sumoylatedhnrnpa2b1controlsthesortingofmirnasintoexosomesthroughbindingtospecificmotifs.nat.commun.4,2980.
wee,l.m.,flores-jasso,c.f.,salomon,w.e.,andzamore,p.d.(2012).argonautedividesitsrnaguideintodomainswithdistinctfunctionsandrna-bindingproperties.cell151,1055–1067.
wyman,s.k.,knouf,e.c.,parkin,r.k.,fritz,b.r.,lin,d.w.,dennis,l.m.,krouse,m.a.,webster,p.j.,andtewari,m.(2011).post-transcriptionalgenerationofmirnavariantsbymultiplenucleotidyltransferasescontributestomirnatranscriptomecomplexity.genomeres.21,1450–1461.
yao,b.,la,l.b.,chen,y.-c.,chang,l.-j.,andchan,e.k.l.(2012).defininganewroleofgw182inmaintainingmirnastability.emborep.13,1102–1108.
zhang,y.,liu,d.,chen,x.,li,j.,li,l.,bian,z.,sun,f.,lu,j.,yin,y.,cai,x.,etal.(2010).secretedmonocyticmir-150enhancestargetedendothelialcellmigration.mol.cell39,133–144.
zhuang,g.,wu,x.,jiang,z.,kasman,i.,yao,j.,guan,y.,oeh,j.,modrusan,z.,bais,c.,sampath,d.,etal.(2012).tumour-secretedmir-9promotesendothelialcellmigrationandangiogenesisbyactivatingthejak-statpathway.emboj.31,3513–3523.
- 还没有人留言评论。精彩留言会获得点赞!