新型GVS系列化合物及其用途的制作方法
本发明属于药物应用领域,具体地,本发明涉及gvs系列化合物及其用途,本发明化合物能够明显改善非酒精性脂肪肝和脂肪堆积。
背景技术:
非酒精性脂肪性肝病(nafld)是指除外酒精和其他明确的损肝因素所致的肝细胞内脂肪过度沉积为主要特征的临床病理综合征,与胰岛素抵抗和遗传易感性密切相关的获得性代谢应激性肝损伤,包括单纯性脂肪肝(sfl)、非酒精性脂肪性肝炎(nash)及其相关肝硬化。随着肥胖及其相关代谢综合征全球化的流行趋势,非酒精性脂肪性肝病现已成为欧美等发达国家和我国富裕地区慢性肝病的重要病因,普通成人nafld患病率10%~30%,其中10%~20%为nash,后者10年内肝硬化发生率高达25%。
非酒精性脂肪性肝病除可直接导致失代偿期肝硬化、肝细胞癌和移植肝复发外,还可影响其他慢性肝病的进展,并参与2型糖尿病和动脉粥样硬化的发病。因此,通过早期防治非酒精性脂肪肝,防止对肝脏进一步的损害极为重要。目前,针对防治非酒精性脂肪肝的药物和保健食品种类很多,但也存在着多方面的问题,如病人不能耐受,药后导致体重增加、体液潴留、心血管等各种不良反应。因此,本领域迫切需要研发出防治非酒精性脂肪肝的新型药物。
技术实现要素:
本发明的目的在于提供一种新型gvs系列化合物及其应用。
本发明第一方面,提供一种式a所示的化合物、其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐的用途,用于制备药物或制剂,所述药物或制剂用于:
(1)治疗和/或预防脂肪肝和脂肪堆积;
(2)降低血清中谷草转氨酶(ast)的活性;
(3)降低血清中谷丙转氨酶(alt)的活性;
(4)降低血清中丙二醛(mda)的水平;
(5)升高超氧化物歧化酶(sod)的活性;
(6)升高血清中脂联素(adp)的水平;
(7)降低脂肪酸合成酶(fas)的活性;
(8)降低低密度脂蛋白胆固醇(ldl-c)的水平;
(9)降低甘油三酯(tg)的水平;和/或
(10)升高高密度脂蛋白胆固醇(hdl-c)的水平;
式中,
x为o或n;
r1为-l-ra,其中l为无,或选自下组的二价连接基团:-(ch2)m-、-(cr4h)m-,m为1、2或3,r4为c1-c4烷基、c1-c4卤代烷基、c3-c4环烷基;
ra选自下组:取代或未取代的c1-c10烷基、取代或未取代的c3-c10环烷基、取代或未取代的含有1-5个选自n、s和o中的杂原子的4-20元杂环基c1-c10烷基、取代或未取代的c5-c20芳基、取代或未取代的c5-c20芳基c1-c10烷基、和取代或未取代的含有1-5个选自n、s和o中的杂原子的c5-c20杂芳基;
所述的取代基选自下组:c1-c10烷基、c1-c6烷基羰基、羧基、硝基和卤素;
r2选自下组:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基和氰基;
r3为一个或多个选自下组的基团:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基、氰基、和3,4-环丁二烯基;
并且,当x为o时,虚线为无,即
在另一优选例中,所述脂肪肝为非酒精性脂肪肝。
在另一优选例中,所述超氧化物歧化酶为血清中超氧化物歧化酶。
在另一优选例中,所述脂肪酸合成酶为血清中脂肪酸合成酶。
在另一优选例中,所述药物或制剂具有选自下组的一个或多个特性:
(a)对pparγ弱激活能力;
(b)对pparγ强结合能力;
(c)弱的前脂肪细胞3t3-l1向脂肪细胞转化的能力。
在另一优选例中,所述“弱激活能力”指测试组测量值m1与对照组参比值m0之比(m1/m0)<0.75,较佳地<0.6,更佳地<0.5,更佳地<0.4,最佳地<0.3,其中,所述对照组的化合物为罗格列酮。
在另一优选例中,所述“强结合能力”指测试组测量值c1与对照组参比值c0之比(c1/c0)>0.9,较佳地>1,更佳地>1.15,更佳地>1.25,最佳地>1.35,其中,所述对照组的化合物为罗格列酮。
在另一优选例中,所述“弱的前脂肪细胞3t3-l1向脂肪细胞转化的能力”指测试组测量值t1与对照组参比值t0之比(t1/t0)<0.5,较佳地<0.25,更佳地<0.1,更佳地<0.06,其中,所述对照组的化合物为罗格列酮。
本发明第二方面,提供一种式a所示的化合物、其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐,
式中,
x为o或n;
r1为-l-ra,其中l为无,或选自下组的二价连接基团:-(ch2)m-、-(cr4h)m-,m为1、2或3,r4为c1-c4烷基、c1-c4卤代烷基、c3-c4环烷基;
ra选自下组:取代或未取代的c1-c10烷基、取代或未取代的c3-c10环烷基、取代或未取代的含有1-5个选自n、s和o中的杂原子的4-20元杂环基c1-c10烷基、取代或未取代的c5-c20芳基、取代或未取代的c5-c20芳基c1-c10烷基、和取代或未取代的含有1-5个选自n、s和o中的杂原子的c5-c20杂芳基;
所述的取代基选自下组:c1-c10烷基、c1-c6烷基羰基、羧基、硝基和卤素;
r2选自下组:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基和氰基;
r3为一个或多个选自下组的基团:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基、氰基、和3,4-环丁二烯基;
并且,当x为o时,虚线为无,即
在另一优选例中,当x为o时,式a化合物为
在另一优选例中,当x为n时,式a化合物为
在另一优选例中,r1选自下组:取代或未取代的c1-c6烷基、取代或未取代的c3-c6环烷基、取代或未取代的含有1-3个选自n、s和o中的杂原子的4-10元杂环基c1-c6烷基、取代或未取代的c5-c10芳基、取代或未取代的c5-c10芳基c2-c6烷基、和取代或未取代的含有1-3个选自n、s和o中的杂原子的5-10元杂芳基。
在另一优选例中,r1选自下组:苄基、苯基、吡啶-4-基甲基、吡啶-4-基乙基、1-苯乙基、甲氧基羰基苄基、和羧基苄基。
在另一优选例中,式a化合物选自下组:
本发明第三方面,提供一种药物组合物,包括:
(i)本发明第一方面和第二方面所述的式a化合物,其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐;和
(ii)药学上可接受的载体。
在另一优选例中,所述的药物组合物含有治疗有效量或安全有效量的式a化合物,其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐。
在另一优选例中,所述药物组合物中,含有0.0001-99wt%(较佳地0.01-90wt%,更佳地,0.1-80wt%)的组分(i),以药物组合物的总重量计。
在另一优选例中,所述的药物组合物用于制备治疗和/或预防脂肪肝的药物。
本发明第四方面,提供一种用于治疗和/或预防脂肪肝的方法,所述方法包括:向所需对象施用如本发明第一方面和第二方面所述的式a化合物或本发明第三方面所述的药物组合物。
在另一优选例中,所述对象包括人。
在另一优选例中,所述对象包括非人哺乳动物。
在另一优选例中,所述非人哺乳动物包括啮齿动物,如小鼠、大鼠。
在另一优选例中,所述脂肪肝会非酒精性脂肪肝。
在另一优选例中,施用剂量为10-10000mg/kg/天,较佳地,500-10000mg/kg/天,更佳地,1000-10000mg/kg/天。
在另一优选例中,施用频率为1-5次/天,较佳地,1-2次/天。
在另一优选例中,施用包括一个或多个周期,各周期为2-30天,较佳地,3-7天。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1显示了化合物gvs-12靶向pparγ活性测试结果.(a)gvs-12的化学结构式;(b)利用cos-7细胞进行荧光素酶报告基因测试gvs-12的pparγ激活作用(n=3,±sem);(c)油红o染色测试;(d)3t3-l1前脂肪细胞中,dmso(1μm)促脂肪分化能力结果;(e)3t3-l1前脂肪细胞中,gvs-12(1μm)促脂肪分化能力结果;(f)3t3-l1前脂肪细胞中,罗格列酮(1μm)促脂肪分化能力结果;。
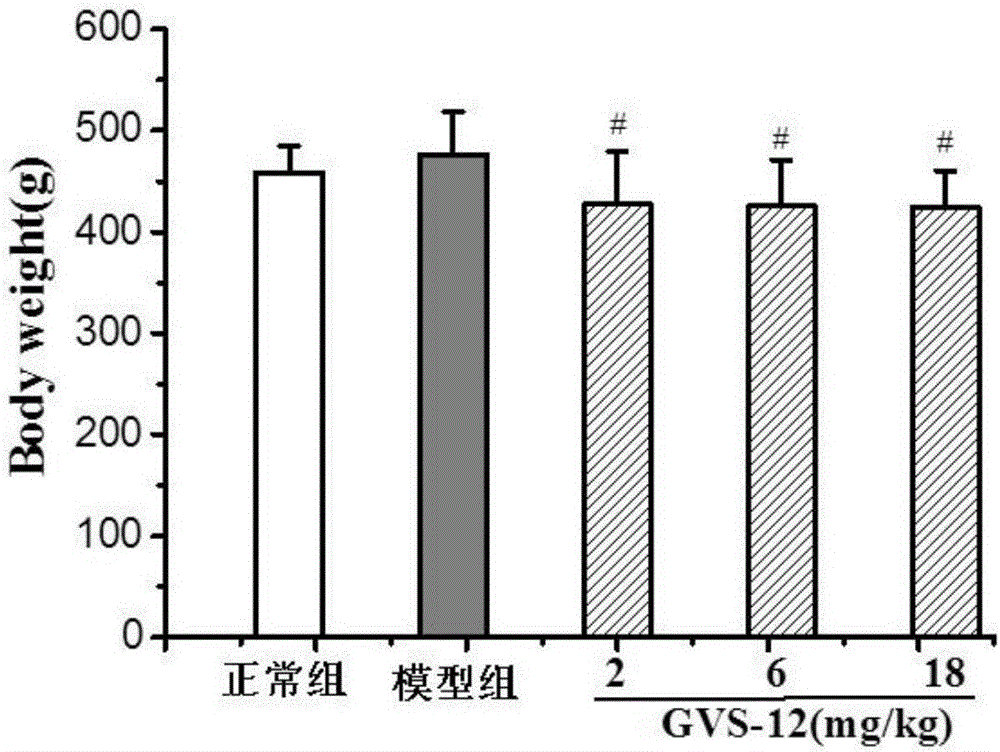
图2显示了化合物gvs-12对脂肪肝大鼠体重的影响#p<0.05vs模型组。
图3显示了化合物gvs-12对非酒精性脂肪肝大鼠的病理学检测。
图4显示了化合物gvs-12对非酒精性脂肪肝大鼠血清中ast、alt、mda、sod的影响(mean±s.e.m)n=10
*p<0.05,**p<0.01vs空白对照组;#p<0.05,##p<0.01vs模型组。
图5显示了化合物gvs-12对非酒精性脂肪肝大鼠血清中ldl-c、hdl-c、tg的影响(mean±s.e.m)n=10
*p<0.05,**p<0.01vs空白对照组;#p<0.05,##p<0.01vs模型组。
图6显示了化合物gvs-12对非酒精性脂肪肝大鼠血清中adp和fas的影响(mean±s.e.m)n=10
*p<0.05,**p<0.01vs空白对照组;#p<0.05,##p<0.01vs模型组。
具体实施方式
本发明人通过广泛而深入的研究,合成了一系列的gvs化合物,所述化合 物能够有效激动pparγ受体,且不明显诱导脂肪分化,在维持正常体重的情况下,具有改善非酒精性脂肪肝和脂肪堆积的作用。在此基础上完成了本发明。
术语说明
除非另外定义,否则本文中所用的全部技术与科学术语均具有如本发明所属领域的普通技术人员通常理解的相同含义。
如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由…构成”、或“由…构成”。
基团定义
如本文所用,术语“取代或未取代的”指所述基团可以是未取代的,或者所述基团中的h被一个或多个(如1-10个,较佳地1-5个,更佳地1-3个,最佳地1-2个)取代基所取代。
如本文所用,所述的“取代”或“取代的”指所述基团具有一个或多个(较佳地1-6个,更佳地1-3个)选自下组的取代基:c1-c6烷基羰基、羧基、硝基和卤素。
如本文所用,术语“c1-c10烷基”是指具有1-10个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、或类似基团;“c1-c3烷基”是指具有1-3个碳原子的直链或支链烷基,例如甲基、乙基、正丙基、异丙基、或类似基团。
如本文所用,术语“c3-c10环烷基”指具有3-10个碳原子的环状烷基,例如环丙基、环丁基、环戊基、环己基、环庚基、或类似基团。
如本文所用,术语“c5-c20芳基”指5至20个(较佳5-14个)碳原子的单价芳香族碳环基团,它具有单环(如苯基)或稠环(如萘基或蒽基),如果连接点在芳香碳原上,稠环可能是非芳香性的(如2-苯并噁唑酮,2h-1,4-苯并噁嗪-3(4h)-酮-7-基等)。优选的芳基包括苯基和萘基。该术语包括取代或未取代的形式,其中取代基的定义如上。
如本文所用,术语“c5-c20杂芳基”指具有5至20个碳原子和1至5个(优选1-3个)选自氧、氮和硫的杂原子的芳香基团,这样的杂芳基可以是单环的(如吡 啶基或呋喃基)或稠环(如吲嗪基(indolizinyl)或苯并噻吩基),其中,所述稠环可以是非芳香性的和/或含有一个杂原子,只要连接点是通过芳香性杂芳基的原子。优选地,杂芳基包括吡啶基、吡咯基、吲哚基、噻吩基和呋喃基。该术语包括取代或未取代的杂芳基。
如本文所用,术语“c4-c20杂环基”指饱和的、部分饱和的或不饱和的基团(但不是芳香性的),具有单环或稠环(包括桥环体系和螺环体系,具有4至20个碳原子和1至4个选自氮、硫或氧的杂原子,在稠环体系中,一个或多个环可以是环烷基、芳基或杂芳基,只要连接点通过非芳香性环。该术语包括取代或未取代的杂环基。
如本文所用,术语“卤素”是指氟、氯、溴、或碘,优选氟、氯和溴。
如本文所用,术语“卤代的”指被相同或不同的一个或多个上述卤原子取代的基团,可以部分卤代或全部卤代,例如三氟甲基、五氟乙基、七氟异丙基、或类似基团。
本发明的化合物可以含有一个或多个不对称中心,并因此以消旋体、外消旋混合物、单一对映体、非对映异构体化合物和单一非对映体的形式出现。可以存在的不对称中心,取决于分子上各种取代基的性质。每个这种不对称中心将独立地产生两个旋光异构体,并且所有可能的旋光异构体和非对映体混合物和纯或部分纯的化合物包括在本发明的范围之内。本发明包括化合物的所有异构形式。
本发明化合物的用途
本发明提供了所述的式a化合物的用途,用于制备药物或制剂,所述药物或制剂用于:
(1)治疗和/或预防脂肪肝和脂肪堆积;
(2)降低血清中谷草转氨酶(ast)的活性;
(3)降低血清中谷丙转氨酶(alt)的活性;
(4)降低血清中丙二醛(mda)的水平;
(5)升高超氧化物歧化酶(sod)的活性;
(6)升高血清中脂联素(adp)的水平;
(7)降低脂肪酸合成酶(fas)的活性;
(8)降低低密度脂蛋白胆固醇(ldl-c)的水平;
(9)降低甘油三酯(tg)的水平;和/或
(10)升高高密度脂蛋白胆固醇(hdl-c)的水平。
本发明化合物
如本文所用,“本发明化合物”、“式a化合物”、“gvs系列化合物”可互换使用,指式a或式a-1或式a-2化合物、其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐。
本发明提供一种式a所示的化合物、其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐,
式中,
x为o或n;
r1为-l-ra,其中l为无,或选自下组的二价连接基团:-(ch2)m-、-(cr4h)m-,m为1、2或3,r4为c1-c4烷基、c1-c4卤代烷基、c3-c4环烷基;
ra选自下组:取代或未取代的c1-c10烷基、取代或未取代的c3-c10环烷基、取代或未取代的含有1-5个选自n、s和o中的杂原子的4-20元杂环基c1-c10烷基、取代或未取代的c5-c20芳基、取代或未取代的c5-c20芳基c1-c10烷基、和取代或未取代的含有1-5个选自n、s和o中的杂原子的c5-c20杂芳基;
所述的取代基选自下组:c1-c10烷基、c1-c6烷基羰基、羧基、硝基和卤素;
r2选自下组:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基和氰基;
r3为一个或多个选自下组的基团:h、卤素、甲基、甲氧基、甲酰胺基、硝基、三氟甲基、氰基、和3,4-环丁二烯基;
并且,当x为o时,虚线为无,即
在另一优选例中,当x为o时,式a化合物为
在另一优选例中,当x为n时,式a化合物为
在另一优选例中,r1选自下组:取代或未取代的c1-c6烷基、取代或未取代的c3-c6环烷基、取代或未取代的含有1-3个选自n、s和o中的杂原子的4-10元杂环基c1-c6烷基、取代或未取代的c5-c10芳基、取代或未取代的c5-c10芳基c2-c6烷基、和取代或未取代的含有1-3个选自n、s和o中的杂原子的5-10元杂芳基。
在另一优选例中,r1选自下组:苄基、苯基、吡啶-4-基甲基、吡啶-4-基乙基、1-苯乙基、甲氧基羰基苄基、和羧基苄基。
在另一优选例中,所述式a化合物为本发明实施例制备的化合物。
药物组合物
本发明还提供了一种药物组合物,它包含安全有效量范围内的活性成分,以及药学上可接受的载体。
本发明所述的“活性成分”是指本发明所述的通式a化合物、其光学异构体、水合物、溶剂化物、其前药或其药学上可接受的盐。
本发明所述的“活性成分”和药物组合物用于制备治疗和/或预防脂肪肝的药物。
“安全有效量”指的是:活性成分的量足以明显改善病情,而不至于产生严重的副作用。通常,药物组合物含有1-2000mg活性成分/剂,更佳地,含有10-200mg活性成分/剂。较佳地,所述的“一剂”为一个药片。
“药学上可接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的活性成分以及它们之间相互掺和,而不明显降低活性成分的药效。
本发明优选实施例的化合物可以作为单独活性药剂给药,也可以与一个或多个其它用于治疗糖尿病的试剂组合使用。本发明优选实施例的化合物与已知的治疗剂组合使用也是有效的,目前已知的化合物和其它治疗糖尿病的治疗剂的组合在优选实施例范围之内。基于药物的特殊性质和所涉及的疾病,本领域普通技术人员能够辨别有效的药剂组合。这种治疗糖尿病的治疗剂包括(但不限于)如下:文迪亚、艾可托等。优选实施例的化合物与治疗糖尿病的治疗剂同时 施用时也有效。
通常,优选实施例的化合物将以治疗有效量、通过具有类似作用的药剂的任意一种可接受的模式施用。优选实施例的化合物(即活性成分)的实际用量根据多个因素确定,如待治疗疾病的严重程度、患者的年龄和相对健康程度、被使用化合物的效力、施用的路径和形式,以及其他因素。该药物可一天施用多次,优选地,每天一次或两次。所有这些因素都在主治医生的考虑范围内。
在本发明中,治疗有效剂量通常可以是对患者一次性施用或分次施用的每日总剂量,例如,每日约0.001至约1000毫克/公斤体重,优选地,每日约1.0至约30毫克/千克体重。单位剂量组合物(dosageunitcomposition)可包含其剂量因数以形成每日剂量。剂型的选择取决于各种因素,例如给药模式和药物物质的生物利用度。通常,优选实施例的化合物可作为药物组合物通过以下任意一种路线给药:口服、全身给药(如透皮、鼻内或通过栓剂)、或肠外给药(如肌内、静脉内或皮下)。优选的给药方式为口服,可根据苦的程度调节方便的日剂量。组合物可采取的形式为片剂、丸剂、胶囊、半固体、粉剂、缓释制剂、溶液、悬浮液、酏剂、气雾剂或任何其他适当的组合物。另一种优选的施用优选实施例化合物的方式为吸入。这是一种将治疗剂直接运送给呼吸道的有效方法(参见,如美国专利号5,607,915)。
合适的药学上可接受的载体或赋形剂包括:如处理剂和药物运送改性剂和促进剂,诸如磷酸钙、硬脂酸镁、滑石、单糖、二糖、淀粉、明胶、纤维素、甲基纤维素钠、羧甲基纤维素、葡萄糖、羟丙基-b-环糊精、聚乙烯吡咯烷酮、低熔点蜡、离子交换树脂等,及其任意两种或多种的组合。液体和半固体的赋形剂可以选自甘油、丙二醇、水、乙醇和各种油,包括石油、动物油、植物油或合成来源,如花生油、豆油、矿物油、芝麻油等。优选的液体载体,特别是用于可注射溶液的载体,包括水、盐水、葡萄糖水性溶液和乙二醇。其它适宜的药学上可接受的赋形剂在《雷明顿药物科学》(remington'spharmaceuticalsciences),mackpub.co.,新泽西(1991)有描述,通过引用纳入本文。
如本文所用,术语“药学上可接受的盐”是指通式i化合物的非毒性酸或碱土金属盐。这些盐可在最终分离和纯化通式i化合物时原位制得、或分别将合适的有机或无机酸或碱与碱性或酸性官能团反应制得。代表性的盐包括,但不限于:乙酸盐、己二酸盐、藻酸盐、柠檬酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、硫酸氢盐、丁酸盐、樟脑酸盐、樟脑磺酸盐、二葡糖酸盐、环戊烷丙酸盐、十二烷基硫酸盐、乙磺酸盐、葡萄糖庚酸盐、甘油磷酸盐、半硫酸盐、庚酸盐、 己酸盐、富马酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、2-羟基乙磺酸盐、乳酸盐、马来酸盐、甲磺酸盐、烟酸盐、2-萘基磺酸盐、草酸盐、双羟萘酸盐、果胶酸盐、硫氰酸盐、3-苯基丙酸盐、苦味酸盐、新戊酸盐、丙酸盐、琥珀酸盐、硫酸盐、酒石酸盐、硫氰酸盐、对甲苯磺酸盐和十一烷酸盐。此外,含氮的碱性基团可被如下试剂季铵盐化:烷基卤化物,如甲基、乙基、丙基、丁基的氯化物、溴化物和碘化物;二烷基硫酸盐,如二甲基、二乙基、二丁基和二戊基硫酸酯;长链卤化物如癸基、月桂基、肉豆蔻基和硬脂基的氯化物、溴化物和碘化物;芳烷基卤化物如苄基和苯乙基溴化物等。由此得到水溶性或油溶性或可分散产品。可被用于形成药学上可接受的酸加成盐的酸的例子包括如盐酸、硫酸、磷酸的无机酸,和如草酸、马来酸、甲磺酸、琥珀酸、柠檬酸的有机酸。碱加成盐可在最终分离和纯化通式i的化合物时原位制得、或使羧酸部分分别与合适的碱(如药学上可接受的金属阳离子的氢氧化物,碳酸盐或碳酸氢盐)或氨、或有机伯、仲或叔胺反应制得。药学上可接受的盐包括,但不限于,基于碱金属和碱土金属的阳离子,如钠、锂、钾、钙、镁、铝的盐等,以及无毒的铵、季铵和胺阳离子,包括,但不限于:铵、四甲基铵、四乙基铵、甲胺、二甲胺、三甲胺、三乙胺、乙胺等。其它代表性的用于形成碱加成盐的有机胺包括二乙胺、乙二胺、乙醇胺、二乙醇胺、哌嗪等。
如本文所用,术语“药学上可接受的前药”是指那些优选实施例的化合物的前药,在体内迅速转化为上述通式所示的母体化合物的化合物,例如在血液中水解。在“t.higuchi和v.stella,作为新型运送系统的前药(pro-drugsasnoveldeliverysystems),a.c.s.15symposiumseries的14卷”和“edwardb.roche编,药物设计中的生物可逆载体(bioreversiblecarriersindrugdesign),美国药学协会和pergamon出版社,1987年”中提供了完整的讨论,这两者都引入本文作为参考。
本发明的主要优点在于:
(1)本发明的化合物可以有效预防和/或治疗脂肪肝,尤其地,在维持正常体重的情况下,具有改善非酒精性脂肪肝和脂肪堆积的作用。
(2)提供一种结构新颖的通式a化合物。
(3)本发明化合物能激动pparγ受体,并且不明显诱导脂肪分化。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说 明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
以下实施例中所用的实验材料和试剂如无特别说明均可从市售渠道获得。
仪器及通用方法
4-羟基苯甲酸、苄溴、苄胺等所涉及化学试剂购于sigma公司。
石油醚、乙酸乙酯等溶剂购于上海国药集团。
brukeram-400型和varianmercuryplus-400型核磁共振仪。
200-300目柱层析硅胶(青岛海洋化工厂)。
hsgf254tlc板(烟台市化工研究院)。
所有溶剂均为分析纯。
制备实施例
实施例1:gvs-1的合成:
步骤1:将对羟基苯甲酸(1.38g,10mmol)和苄胺(1.08g,10mmol)置于25ml圆底烧瓶中,加入bop缩合剂(5.3g,12mmol)和dmap(1.46g,12mmol),后加入干燥的dmf20ml,室温搅拌,tlc跟踪反应,3h后反应完全ea萃取水洗浓缩得到中间体1,ms(ei):m/z227.1[m+h]+.
步骤2:将中间体1(230mg,1mmol)和3,4-二氟溴化苄(206mg,1mmol)置于25ml圆底烧瓶中,加入cs2co3(490mg,1.5mmol)于烧瓶中,后加入干燥的dmf8ml,室温搅拌,tlc跟踪反应,3h后反应完全,用水/乙酸乙酯萃取,直至水相没有化合物后,合并有机相用水洗涤两次,再用饱和nacl溶液洗涤,无水硫酸钠干燥,最后硅胶柱分离纯化(石油醚/乙酸乙酯=8:1)得到目标产物。收率80%,产物为白色固体。1hnmr(400mhz,氯仿-d)δ7.79–7.73(m,2h),7.44–7.37(m,1h),7.35(d,j=4.0hz,4h),7.32–7.27(m,1h),7.25–7.21(m,1h),7.14(m,1h),6.99–6.92(m,2h),6.38(t,j=5.7hz,1h),5.06(t,j=0.7hz,2h), 4.63(d,j=5.6hz,2h).lcms(esi):m/z370.1[m+h]+.
实施例2:gvs-2的合成:
用3-氟-4-氯溴化苄代替3,4-二氟溴化苄照制备gvs-1的方法制备gvs-2(产率85%)。1hnmr(400mhz,氯仿-d)δ7.80–7.73(m,2h),7.36(d,j=4.4hz,4h),7.33–7.25(m,2h),7.25–7.10(m,2h),7.01–6.93(m,2h),6.32(t,j=5.4hz,1h),5.05(s,2h),4.64(d,j=5.6hz,2h).lcms(esi):m/z354.1[m+h]+.
实施例3:gvs-3的合成:
用4-三氟甲基溴化苄代替3,4-二氟溴化苄照制备gvs-1的方法制备gvs-3(产率81%)。1hnmr(400mhz,氯仿-d)δ7.80–7.74(m,2h),7.69–7.61(m,2h),7.54(m,2h),7.35(d,j=4.6hz,4h),7.33–7.27(m,1h),7.02–6.95(m,2h),6.34(s,1h),5.17(s,2h),4.63(d,j=5.6hz,2h).lcms(esi):m/z386.1[m+h]+.
实施例4:gvs-4的合成:
步骤1:用4-甲氨基吡啶代替苄胺照制备中间体1的方法制备中间体2,lcms(esi):m/z229.1[m+h]+.
步骤2:用中间体2代替中间体1按照制备gvs-1的方法制备gvs-4(产率79%)。1hnmr(400mhz,氯仿-d)δ8.55(d,j=4.7hz,2h),7.79(d,j=8.4hz,2h),7.41(t,j=7.8hz,1h),7.25(s,2h),7.18–7.10(m,1h),6.98(d,j=8.4hz,2h),6.60(t,j=6.1hz,1h),5.08(s,2h),4.64(d,j=6.0hz,2h).lcms(esi):m/z355.1[m+h]+.
实施例5:gvs-5的合成:
用中间体2代替中间体1按照制备gvs-2的方法制备gvs-5(产率75%)。1hnmr(400mhz,氯仿-d)δ8.52(d,j=5.2hz,2h),7.80(d,j=8.4hz,2h),7.31–7.11(m,4h),6.97(d,j=8.4hz,2h),6.78(d,j=6.2hz,1h),5.05(s,2h),4.62(d,j=6.0hz,2h).lcms(esi):m/z371.1[m+h]+.
实施例6:gvs-6的合成:
用中间体2代替中间体1按照制备gvs-3的方法制备gvs-6(产率77%)。1hnmr(400mhz,氯仿-d)δ8.59–8.51(m,2h),7.83–7.77(m,2h),7.65(d,j=8.1hz,2h),7.58–7.52(m,2h),7.25–7.22(m,2h),7.04–6.96(m,2h),6.63(t,j=6.0hz,1h),5.17(s,2h),4.64(d,j=6.0hz,2h).lcms(esi):m/z387.1[m+h]+.
实施例7:gvs-7的合成:
步骤1:用3-甲氨基吡啶代替苄胺照制备中间体1的方法制备中间体3,lcms(esi):m/z229.1[m+h]+.
步骤2:用中间体3代替中间体1按照制备gvs-1的方法制备gvs-7(产率81%)。1hnmr(400mhz,氯仿-d)δ8.63–8.56(m,1h),8.53(dd,j=5.0,1.7hz,1h),7.79–7.74(m,2h),7.71(d,j=7.9hz,1h),7.40(t,j=7.8hz,1h),7.30–7.27(m,1h),7.23(dd,j=9.7,2.0hz,1h),7.16–7.12(m,1h),6.99–6.94(m,2h),6.53(t,j=5.9hz,1h),5.07(s,2h),4.64(d,j=5.9hz,2h).lcms(esi):m/z355.1[m+h]+.
实施例8:gvs-8的合成:
用中间体3代替中间体1按照制备gvs-2的方法制备gvs-8(产率70%)。1hnmr(400mhz,氯仿-d)δ8.58(d,j=2.3hz,1h),8.53(dd,j=4.9,1.7hz,1h),7.83–7.73(m,2h),7.71(dt,j=7.9,2.0hz,1h),7.40(t,j=7.8hz,1h),7.30–7.20(m,3h),7.18–7.10(m,1h),7.02–6.91(m,2h),6.53(t,j=6.1hz,1h),5.07(s,2h),4.64(d,j=5.9hz,2h).lcms(esi):m/z371.1[m+h]+.
实施例9:gvs-9的合成:
用中间体3代替中间体1按照制备gvs-3的方法制备gvs-9(产率78%)。1hnmr(400mhz,氯仿-d)δ8.49(dd,j=17.4,3.8hz,2h),7.81–7.75(m,2h),7.68(dd,j=7.8,2.0hz,1h),7.66–7.60(m,2h),7.52(d,j=8.0hz,2h),7.23(dd,j=7.8,4.9hz,1h),6.95(dt,j=9.6,2.5hz,3h),5.14(s,2h),4.59(dd,j=6.0,2.8hz,2h).lcms(esi):m/z387.1[m+h]+.
实施例10:gvs-10的合成:
步骤1:用3-甲氨基呋喃代替苄胺照制备中间体1的方法制备中间体4,lcms(esi):m/z218.1[m+h]+.
步骤2:用中间体4代替中间体1按照制备gvs-1的方法制备gvs-10(产率85%)。1hnmr(400mhz,氯仿-d)δ8.12(dd,j=1.8,0.7hz,1h),7.65(dd,j=8.6,1.8hz,1h),7.38(dd,j=1.8,0.9hz,1h),7.23(dd,j=8.7,0.8hz,1h),7.16(d,j=3.2hz,1h),7.08(m,1h),6.63(dd,j=3.3,0.8hz,1h),6.46(t,j=5.2hz,1h),6.37–6.26(m,2h),5.29(d,j=1.3hz,2h),4.67(dd,j=5.5,0.8hz,2h).lcms(esi):m/z344.1[m+h]+.
实施例11:gvs-11的合成:
用中间体4代替中间体1按照制备gvs-2的方法制备gvs-11(产率79%)。1hnmr(400mhz,氯仿-d)δ7.79–7.70(m,2h),7.44–7.35(m,2h),7.23(dd,j=9.7,1.9hz,1h),7.14(ddt,j=8.3,2.1,0.9hz,1h),6.99–6.92(m,2h),6.38–6.31(m,2h),6.31–6.27(m,1h),5.06(s,2h),4.62(d,j=5.4hz,2h).lcms(esi):m/z360.1[m+h]+.
实施例12:gvs-12的合成:
用中间体4代替中间体1按照制备gvs-3的方法制备gvs-12(产率82%)。1hnmr(400mhz,氯仿-d)δ7.81–7.72(m,2h),7.65(d,j=8.0hz,2h),7.58–7.52(m,2h),7.37(dd,j=1.9,0.9hz,1h),7.01–6.95(m,2h),6.36–6.32(m,2h),6.29(m,1h),5.16(s,2h),4.66–4.59(m,2h).lcms(esi):m/z376.1[m+h]+.
实施例13:gvs-13的合成:
向反应瓶中加入吲哚-5-甲酸甲酯(0.2mmol)、3-氟,4-氯苄溴(0.22mmol)、nah(0.22mmol)和dmf(n,n-二甲基甲酰胺)(2ml),冰浴下搅拌直至室温,tlc检测反应完全。向反应瓶中加入1mol/l盐酸(50ml),乙酸乙酯萃取3次,合并有机相。再用1mol/l盐酸、饱和nacl各洗有机相1次。无水硫酸钠干燥过夜。抽滤除去干燥剂,旋干得粗产品。再经硅胶柱色谱分离纯化,得1-(4-氯-3-氟苄基)-1h-吲哚-5-羧酸甲酯(b-1)(57mg,0.18mmol),产率90%。
向所得1-(4-氯-3-氟苄基)-1h-吲哚-5-羧酸甲酯(b-1)(0.18mmol)中加入4mol/l的naoh溶液(8ml),回流搅拌,tlc检测反应完全。向反应瓶中加入1mol/l盐酸溶液调ph为酸性,乙酸乙酯萃取3次,合并有机相。再用1mol/l盐 酸、饱和nacl各洗有机相1次。无水硫酸钠干燥过夜。抽滤除去干燥剂,旋干得粗产品。再经硅胶柱色谱分离纯化,得水解产物1-(4-氯-3-氟苄基)-1h-吲哚-5-羧酸(b-2)(52mg,0.17mmol),产率95%。
向所得1-(4-氯-3-氟苄基)-1h-吲哚-5-羧酸(b-2)(0.17mmol)加入苄胺(0.19mmol)、bop(苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐)(0.34mmol)、dmap(4-二甲氨基吡啶)(0.34mmol)和dmf(n,n-二甲基甲酰胺)(2ml),室温搅拌,tlc检测反应完全。向反应瓶中加入1mol/l盐酸(50ml),乙酸乙酯萃取3次,合并有机相。再用1mol/l盐酸、饱和nacl各洗有机相1次。无水硫酸钠干燥过夜。抽滤除去干燥剂,旋干得粗产品。再经硅胶柱色谱分离纯化,得缩合产物n-苄基-1-(4-氯-3-氟苄基)-1h-吲哚-5-甲酰胺(gvs-13)(63mg,0.16mmol),产率92%。1hnmr(400mhz,dmso):δ8.92(s,1h),8.19(s,1h),7.75–7.65(m,1h),7.62(d,1h,j=3.2hz),7.56–7.48(m,2h),7.32(s,2h),7.31(s,2h),7.27(dd,1h,j=10.3,1.8hz),7.24-7.20(m,1h),7.00(dd,1h,j=8.3,1.4hz),6.62(d,1h,j=3.1hz),5.49(s,2h),4.49(d,2h,j=6.0hz).
实施例14:gvs-14的合成:
用苯胺代替苄胺按照制备gvs-13的方法制备gvs-14(产率91%)。1hnmr(400mhz,dmso):10.13(s,1h),8.26(d,1h,j=1.0hz),7.83-7.77(m,2h),7.74(dd,1h,j=8.6,1.5hz),7.66(d,1h,j=3.2hz),7.59(d,1h,j=8.7hz),7.54(t,1h,j=8.1hz),7.34(t,2h,j=7.9hz),7.29(dd,1h,j=10.2,1.5hz),7.07(t,1h,j=7.4hz),7.01(d,1h,j=7.8hz),6.68(d,1h,j=3.1hz),5.52(s,2h).
实施例15:gvs-15的合成:
用4-胺甲基吡啶代替苄胺按照制备gvs-13的方法制备gvs-15(产率93%)。1hnmr(400mhz,dmso):δ9.01(t,1h,j=5.9hz),8.49(dd,2h,j=4.5,1.5hz),8.21(s,1h),7.70(dd,1h,j=8.7,1.5hz),7.63(d,1h,j=3.2hz),7.59–7.46(m,2h), 7.30(d,2h,j=5.9hz),7.29–7.25(m,1h),7.07–6.94(m,1h),6.64(d,1h,j=3.0hz),5.49(s,2h),4.50(d,2h,j=5.9hz).
实施例16:gvs-16的合成:
用1-苯乙基乙胺代替苄胺按照制备gvs-13的方法制备gvs-16(产率92%)。1hnmr(400mhz,dmso):δ8.66(d,1h,j=8.1hz),8.19(d,1h,j=1.2hz),7.67(dd,1h,j=8.6,1.6hz),7.61(d,1h,j=3.2hz),7.54-7.50(m,2h),7.41(s,1h),7.39(s,1h),7.31(t,2h,j=7.6hz),7.25(dd,1h,j=10.3,1.7hz),7.20(t,1h,j=7.3hz),6.99(d,1h,j=8.3hz),6.63(d,1h,j=2.9hz),5.48(s,2h),5.22-5.15(m,1h),1.48(d,3h,j=7.1hz).
实施例17:gvs-17的合成:
用3、4-二氟溴化苄代替3-氟-4-氯溴化苄按照合成gvs-13的方法制备gvs-17(产率72%)。1hnmr(400mhz,dmso-d6)δ8.93(s,1h),8.21(s,1h),7.71(d,1h,j=8.7hz),7.62(d,1h,j=3.2hz),7.56(d,1h,j=8.7hz),7.42-7.28(m,6h),7.24-7.20(m,1h),7.09-6.96(m,1h),6.62(d,1h,j=3.1hz),5.46(s,2h),4.50(d,2h,j=5.7hz);hrms(esi)m/zcalcdforc23h18f2n2o[m+na]+:399.1279,found:376.1387
实施例18:gvs-18的合成:
用4-异丙基苄胺代替苄胺按照合成gvs-17的方法制备gvs-18(产率76%)。1hnmr(400mhz,dmso-d6)δ8.90(s,1h),8.22(s,1h),7.78-7.51(m,3h),7.39-7.29(m,2h),7.21(dd,4h,j=32.7and7.8hz),7.03(s,1h),6.62(d,1h,j=2.6hz),5.45(s,2h),4.47(d,2h,j=5.6hz),2.83(t,1h,j=6.8hz),1.16(d,6h, j=6.9hz);hrms(esi)m/zcalcdforc26h24f2n2o[m+na]+:441.1756,found:418.1857.
实施例19:gvs-19的合成:
用2-胺甲基呋喃代替苄胺按照合成gvs-17的方法制备gvs-19(产率79%)。1hnmr(400mhz,dmso-d6)δ8.82(t,1h,j=7.6hz),8.17(d,1h,j=2.0hz),7.67(dd,1h,j=11.6and2.4hz),7.62(d,1h,j=4.4hz),7.57-7.53(m,2h),7.42-7.28(m,2h),7.05-7.00(m,1h),6.61(d,1h,j=4.0hz),6.39(dd,1h,j=4.4and2.4hz),6.25(dd,1h,j=4.4and0.8hz),5.45(s,2h),4.47(d,2h,j=7.6hz);hrms(esi)m/zcalcdforc21h16f2n2o2[m+h]+:367.1254,found:366.1180.
实施例20:gvs-20的合成:
用溴化苄代替3-氟-4-氯溴化苄按照合成gvs-13的方法制备gvs-20(产率82%)。1hnmr(400mhz,氯仿-d)δ8.13(dd,j=1.8,0.7hz,1h),7.64(dd,j=8.6,1.8hz,1h),7.42–7.26(m,9h),7.18(d,j=3.2hz,1h),7.11–7.04(m,2h),6.61(dd,j=3.2,0.9hz,1h),6.47(t,j=5.7hz,1h),5.33(s,2h),4.67(d,j=5.6hz,2h).lcms(esi):m/z341.2[m+h]+.
实施例21:gvs-21的合成:
用2-溴甲基奈代替3-氟-4-氯溴化苄按照合成gvs-13的方法制备gvs-21(产率69%)。1hnmr(400mhz,氯仿-d)δ8.15(dd,j=1.8,0.8hz,1h),7.85–7.68(m,3h),7.63(dd,j=8.6,1.8hz,1h),7.52(s,1h),7.49–7.42(m,2h),7.41–7.26(m,6h),7.25–7.18(m,2h),6.64(dt,j=3.2,0.8hz,1h),6.40(d,j=6.0 hz,1h),5.50(s,2h),4.68(d,j=5.6hz,2h).lcms(esi):m/z391.2[m+h]+.
实施例22:gvs-22的合成:
用4-三氟甲基溴化苄代替3、4-二氟溴化苄按照合成gvs-18的方法制备gvs-22(产率77%)。1hnmr(400mhz,dmso-d6)δ8.87(t,1h,j=6.0hz),8.20(s,1h),7.65(dd,4h,j=23.7and5.5hz),7.49(d,1h,j=8.6hz),7.34(d,2h,j=8.0hz),7.21(dd,4h,j=24.9and8.1hz),6.63(d,1h,j=3.0hz),5.59(s,2h),4.44(d,2h,j=5.8hz),2.93–2.75(m,1h),1.17(d,6h,j=6.9hz);hrms(esi)m/zcalcdforc27h25f3n2o[m+h]+:451.1987,found:450.1919.
体外活性测试
实施例23:化合物对pparγ激活能力评估(荧光素酶活性测定法):
cos-7细胞购自atcc,培养于10%fbs无抗生素dmem,37℃,5%co2孵育箱中。待细胞进入对数生长期接种于24孔板中,细胞融合约70%时,按照lipofectamine2000(invitrogen)操作说明进行质粒共转染(50ng全长hpparγ,100ngpparγ,5ng海肾荧光素酶质粒)。24h后转染细胞分别用1μm化合物干预,其中1μm罗格列酮做阳性对照,dmso做阴性对照。干预24h后按reporterluciferaseassaykits(promega)操作说明测定荧光素酶活性,每组设定3个独立测试孔。
实验结果见表1。
表1luciferase方法测试化合物gvs1-22对pparγ的激活能力
(注:激活能力在0.1-0.5标记为+,在0.5-1.0标记为++)
实施例24:tr-fret方法测试化合物与pparγ结合能力步骤:
1.将化合物1至22用dmso稀释到1mm。dmso做阴性对照,罗格列酮阳性对照。
2.将稀释好的化合物(1至22,罗格列酮)用tr-fretbuffer再次稀释到2μm。
3.以tr-fretbuffer为溶剂准备fluormonetmpan-ppargreensolution(20nm)。
4.以tr-fretbuffer为溶剂准备20nmtbanti-gstantibody和4μmpparγ-lbdprotein。
5.将20μl步骤2溶液,10μl步骤3溶液和10μl步骤4溶液混合在384孔板中,震荡6小时。在酶标仪上读数。
实验结果见表2。
表2tr-fret方法测试化合物gvs1-22与pparγ结合能力
(注:结合能力在0.1-0.5标记为+,在0.5-0.8标记为++,在0.8-1.5标记为+++)
表3部分化合物激活能力与结合能力的比值
(注:比值在0.1-0.5标记为+,在0.5-1.0标记为++,在1.0-5.0标记为+++)
实施例25:gvs化合物致脂肪分化能力测试及real-timepcr测定
3t3-l1前脂肪细胞购自atcc,培养于含青霉素-链霉素双抗的10%fbsdmem,37℃,5%co2孵育箱中。接种于培养板,汇合后2d加入诱导液(10%fbsdmem含0.5mmol/libmx(3-异丁基-1-甲基黄嘌呤),1μmol/ldex(地塞米松),850nmol/l胰岛素)。72h后换成10%fbs高糖dmem含850nmol/l胰岛素,每2d换一次。1μm罗格列酮为阳性对照,dmso为阴性对照,样品组为1μm。诱导开始第8d进行油红o染色及dapi染色,显微镜(olympus)拍照,计算脂肪细胞分化率。
实验结果见表4。
表4gvs化合物致脂肪分化能力
实施例26:gvs系列化合物在非酒精性脂肪肝中的作用
1.材料
1.1动物
动物spf级sd雄性大鼠60只,体重180±200g,由桂林医学院spf实验动物中心提供。
1.2药品与试剂
合成gvs系列化合物(纯度>99.5%),谷丙转氨酶(alt)、谷草转氨酶(ast)、丙二醛(mda)和超氧化物歧化酶(sod)试剂盒购于南京建成生物工程研究所。
低密度脂蛋白胆固醇(ldl-c)、高密度脂蛋白胆固醇(hdl-c)、甘油三酯(tg)试剂盒购于北京北化康泰临床试剂有限公司。
脂联素(adp)和脂肪酸合成酶(fas)购于武汉伊莱瑞特生物科技有限公司。
1.3原料与仪器
h2050r台式冷冻离心机,湖南湘仪离心机仪器有限公司;
epoch酶标仪,美国biotek公司;
752型分光光度计,上海第三分析仪器厂;
o1ympusbx51显微镜,奥林巴斯生产;
auw220d电子天平,德国sartorius公司;
tanon电泳仪,上海天能。
2.方法
2.1分组和模型建立
取sd大鼠50只(体重180±200克)随机分为5组(n=10):
①空白对照组;
②模型组;
③gvs-12低剂量组(2mg/kg);
④gvs-12中剂量组(6mg/kg);
⑤gvs-12高剂量组(18mg/kg)。
建立高脂饮食诱导的非酒精性脂肪肝大鼠模型,空白组给予普通饲料喂养16周;模型组及给药组给予高脂饲料(饲料配比:82.5%普通饲料+10%猪油+5% 蛋黄粉+2%胆固醇+0.5%胆酸钠)喂养16周,并于第四周开始给药,gvs-12皮下注射给药,连续给药12周,空白对照组和模型组给予等体积的蒸馏水。
2.2检测指标
严格按照试剂盒的要求规范操作。
谷丙转氨酶(alt)、谷草转氨酶(ast)、丙二醛(mda)、超氧化物歧化酶(sod)试剂盒购于南京建成生物工程研究所。
低密度脂蛋白胆固醇(ldl-c)、高密度脂蛋白胆固醇(hdl-c)、总胆固醇(tc)、甘油三脂(tg)试剂盒购于北京北化康泰临床试剂有限公司。
脂联素(adp)和脂肪酸合成酶(fas)试剂盒购于武汉伊莱瑞特生物科技有限公司。
2.3病理学观察
取各大鼠肝脏同一部位组织,以10%中性福尔马林固定,脱水,包埋,切片,脱蜡,采用油红o染色,直观检测gvs-12对该模型脂质沉积的改善情况。采用he染色观察大鼠肝脏脂肪堆积程度及炎症程度,镜下观察分析肝脏组织病理情况。
2.4数据处理
用spss13.0软件进行统计学分析。试验数据用平均值±标准差(mean±s.e.m)表示,各组间均数的比较采用单因素方差分析(one-wayanovea),再采用dunnett-t检验,以p<0.05作为显著性差异标准。
3.结果
(1)检测gvs-12对非酒精性脂肪肝大鼠体重的影响。
由图2可知,给药组大鼠的体重与模型组相比,没有显著增加。
(2)病理形态学观察,采用油红o和he染色观察大鼠变性程度及炎症程度。
由图3可知,油红o染色结果表明模型组的脂肪已被显著分化和沉积,而给药组的脂肪分化明显降低。
he染色结果表明模型组肝组织出现严重的炎症损伤和肝纤维化,给药组炎症损伤逐渐减少,且呈剂量依赖性不断地被改善。
(3)通过生化法检测gvs-12对脂肪肝大鼠肝功能因子谷草转氨酶(ast)、谷丙转氨酶(alt)、丙二醛(mda)、超氧化物歧化酶(sod)的影响。
由图4可知,与模型组比相比,gvs-12能明显降低血清中谷草转氨酶(ast)、谷丙转氨酶(alt)、丙二醛(mda)的活性及浓度,同时能够明显升高超氧化物歧化酶(sod)的活性;
(4)通过生化法检测gvs-12对脂肪肝大鼠脂肪因子低密度脂蛋白胆固醇(ldl-c)、高密度脂蛋白胆固醇(hdl-c)、甘油三酯(tg)的影响;
由图5可知,与模型组比相比,gvs-12能降低低密度脂蛋白胆固醇(ldl-c)、甘油三酯(tg)以及增加高密度脂蛋白胆固醇(hdl-c)的含量。
(5)通过elisa法检测gvs-12对脂肪肝大鼠脂联素(adp)、脂肪酸合成酶(fas)的影响。
由图6可知,与模型组比相比,能升高血清中脂联素(adp)的含量和降低脂肪酸合成酶(fas)的活性。
以上结果充分表明gvs-12具有在维持正常体重的情况下,能改善非酒精性肝脏炎症和脂肪堆积的作用,具有良好防治非酒精性脂肪肝的作用。
综上,本发明的具有全新母核结构的gvs系列化合物结构不同于罗格列酮,原料简单易得,能绑定和激动pparγ受体,且具有不明显诱导脂肪分化特点作用,具有与罗格列酮相当甚至更优的结果。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!