烟草中一种异吲哚生物碱化合物、其制备方法和应用与流程
本发明属于烟草化学技术领域,具体涉及一种从烟草中首次提取得到的异吲哚生物碱化合物。同时,本发明还涉及该化合物的制备方法和在抗烟草花叶病毒中的应用。
背景技术:
烟草是世界上化学成分最为复杂的植物,次生代谢产物非常丰富,经过几十年的研究,人们目前从烟草中鉴定出来的单体化学物质就超过3000多种,而且还有许多成分尚未鉴定出来。烟草除主要用于卷烟抽吸用途外,还可从中提取多种有利用价值的化学成分,从中发现有开发利用价值的先导性化合物。吲哚生物碱是指分子中含吲哚环的天然生物碱类化合物。吲哚生物碱类化合物分布较广,在植物体广泛存在;而且许多化合物具有突出的活性,是天然药物和生物农药的重要来源。
技术实现要素:
本发明从云南烤烟烟叶中分离得到了一种新的芳构化异吲哚生物碱化合物,该化合物至今尚未见到相关报道,值得一提的是该化合物具有显著的抗烟草花叶病病毒活性。
本发明的目的在于提供一种新的异吲哚生物碱化合物。
本发明的另一个目的是提供一种制备所述异吲哚生物碱化合物的方法。
本发明的目的还在于提供所述异吲哚生物碱化合物在抗烟草花叶病毒中的应用。
除非另有说明,本发明中所采用的百分数均为质量百分数。
本发明第一方面从烟草中分离出一种新的异吲哚生物碱化合物,其分子式为C13H15NO6,具有下述结构式:
化合物命名为:4,6-二羟基-2-(2-羟乙基)-5-(3-羟基丙酮基)-异吲哚-1-酮;英文名为:4,6-dihydroxy-2-(2-hydroxyethyl)-5-(3-hydroxypropan)-isoindolin-1-one。
本发明第二方面制备所述异吲哚生物碱化合物的方法,该方法包括以下步骤:
(1)浸膏提取:以烟叶为原料,将烟叶粉碎或切成小段,用重量百分浓度80%~100%的甲醇或乙醇,或重量百分浓度60%~90%的丙酮为提取溶剂,提取溶剂:烟叶的重量比=2~4:1,浸泡24h~72h,提取3~5次,合并提取液、过滤浓缩成浸膏;
(2)硅胶柱层析:浸膏用重量比2~4倍量的160~300目硅胶干法装柱进行硅胶柱层析;以氯仿-丙酮溶液进行梯度洗脱,合并相同的部分,收集各部分洗脱液并浓缩;
(3)高压液相色谱分离纯化:洗脱液的7:3部分进一步用高压液相色谱分离纯化即得所需的异吲哚生物碱化合物。
优选地,其还包括以下进一步提纯的步骤:将在所述高压液相色谱分离之后得到的所述的异吲哚生物碱化合物再次溶于纯甲醇,并以纯甲醇为流动相,通过凝胶柱进行层析分离,得到进一步提纯的所述的异吲哚生物碱化合物。
优选地,步骤(3)中,所述的高压液相色谱分离纯化是采用21.2mm×250mm,5μm的C18色谱柱,流速为20mL/min,流动相为36wt%的甲醇,紫外检测器检测波长为298nm,每次进样200μL,收集29.5min的色谱峰,多次累加后蒸干。
优选地,步骤(2)中,所述的浸膏在经硅胶柱层析粗分前,用重量1.5~3倍量的纯甲醇或纯乙醇或纯丙酮溶解后,用重量0.8~1.2倍的80~100目硅胶拌样。
优选地,步骤(2)中,所述的梯度洗脱,氯仿-丙酮溶液体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1和1:2。
本发明第二方面提供所述的异吲哚生物碱化合物在制备抗烟草花叶病毒药物中的应用。
表-1.化合物的1H NMR和13C NMR数据(溶剂为CDCl3)
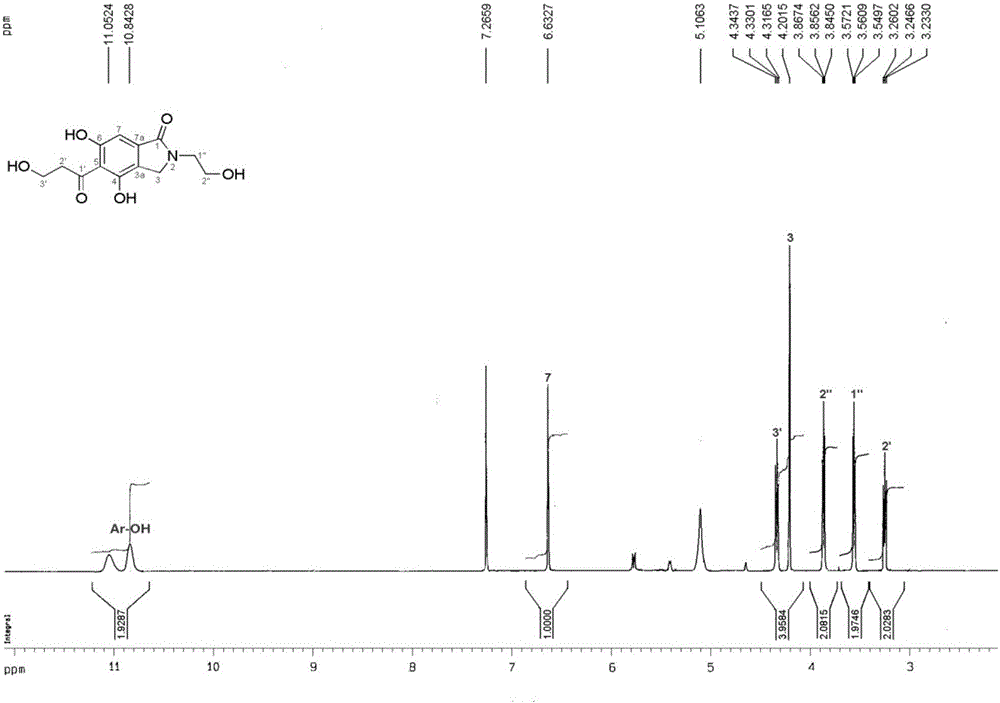
以上述方法制备得到的异吲哚生物碱化合物的结构通过以下方法进行测定。本发明化合物为黄色胶状物,由高分辨质谱可知其准分子离子峰[M+Na]+为304.0790,可推测其分子式为C13H15NO6,化合物的不饱和度为7。紫外光谱在210、260和298nm有吸收峰,说明化合物中有芳环结构。红外光谱在1688,1660,1612,1576,1452cm-1存在强吸收峰,证实化合物中存在羟基、羰基和芳环功能团。根据化合物的1H和13C NMR数据(C-1-C-7a;H2-3,和H-7)(表-1)可初步推测化合物1为异吲哚-1-酮结构。根据H2-3(δH 4.20)和C-1(δC 166.3)、C-3a(δC136.3)、C-4(δC 153.1)、C-7a(δC 116.8);H-7(δH 6.63)和C-1(δC 166.3)、C-3a(δC 136.3)、C-7a(δC 116.8)的HMBC相关(图-3)可进一步证实本发明化合物为异吲哚-1-酮结构。化合物的母核得到确定后,其它基团可归属为取代基,包括一个3-羟基丙酮基(C-1'–C-3',H2-2',H2-3'),一个和氮原子相连的羟乙基(C-1”、C-2”,H2-1”,H2-2”)和两个酚羟基(11.05s、10.84)。根据H2-2'和C-4、C-5和C-6的HMBC相关可证实3-羟基丙酮基取代在苯环的C-5位;羟乙基取代在异吲哚环的氮原子上和由H2-1”和C-1、C-3,以及H2-3和C-1”的HMBC相关确定,根据酚羟基氢(δH 11.05)和C-3a、C-4、C-5,以及(δH 10.84)和C-4、C-5、C-6的HMBC相关可证实两个酚羟基取代在C-4和C-6位。至此,本发明化合物的结构的到确定,并命名为4,6-二羟基-2-(2-羟乙基)-5-(3-羟基丙酮基)-异吲哚-1-酮。
化合物的红外、紫外和质谱数据:UV(MeOH)λmax nm(logε)210(4.18)、268(3.52)、和306(3.08);IR(KBr)νmax 3386、2938、1688、1660、1612、1576、1452、1358、1246、1167、1062、973、和852cm-1;正离子模式ESIMS m/z 304[M+Na]+,正离子模式HRESIMS m/z 304.0790[M+Na]+(计算值为304.0797,C13H15NNaO6,)。
采用半叶法进行了本发明化合物的抗烟草花叶病毒活性测试,结果明本化合物的相对抑制率为56.8%,超过对照宁南霉素的相对抑制率31.5%,说明化合物有很好的抗烟草花叶病毒活性。
本发明的有益效果:
1、本发明化合物是首次被分离出来的,通过上述核磁共振和质谱测定方法确定为异吲哚生物碱化合物,并表征了其具体结构,结构新颖,至今尚未见到相关报道。
2、活性检测结果揭示,本发明的异吲哚生物碱化合物在制备抗轮状病毒药物中有良好的应用前景。本发明化合物结构简单活性较好,可作为抗花叶病毒药物研发的先导性化合物用于抗花叶病毒药物制剂研发。
附图说明
图1为本发明异吲哚生物碱化合物的核磁共振碳谱;
图2为本发明异吲哚生物碱化合物的核磁共振氢谱;
图3为本发明异吲哚生物碱化合物的主要HMBC相关。
具体实施方式
下面结合附图和实施例对本发明作进一步的详细说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或改进,均落入本发明的保护范围。
实施例1
制备异吲哚生物碱化合物C13H15NO6,包括浸膏提取、硅胶柱层析、高压液相色谱分离,具体采用以下步骤:
1.浸膏提取:取烟叶粉碎,用高浓度甲醇(wt%:95%)或高浓度乙醇(wt%:95%)或高浓度丙酮(wt%:70%)为提取溶剂,提取溶剂:烟草(重量比)=3:5,浸泡54h,提取4次,合并提取液、过滤浓缩成浸膏。
2.硅胶柱层析:浸膏用重量2.5倍量的纯甲醇或纯乙醇或纯丙酮溶解后用重量1.2倍的80~100目硅胶拌样,用重量3倍量的250目硅胶干法装柱进行硅胶柱层析;以体积配比为(1:0、20:1、9:1、8:2、7:3、6:4、1:1和1:2)的氯仿-丙酮溶液进行梯度洗脱,合并相同的部分,收集各部分洗脱液并浓缩。
3.高压液相色谱分离:柱层析洗脱液的7:3部分进一步用高压液相色谱分离纯化即得所述的异吲哚生物碱化合物,高压液相色谱分离纯化是采用21.2mm×250mm,5μm的C18色谱柱,流速为20mL/min,流动相为36wt%的甲醇,紫外检测器检测波长为298nm,每次进样200μL,收集29.5min的色谱峰,多次累加后蒸干。
高压液相色谱法分离纯化后的物质,一个优选的后处理方案为,所得化合物再次用纯甲醇溶解,再以纯甲醇为流动相,用凝胶柱层析分离,以进一步分离纯化。
本发明所用烟叶原料不受地区和品种限制,均可以实现本发明,下面以来源于云南中烟工业有限责任公司不同产地的烟叶原料,对本发明做进一步说明:
实施例2
烟草样品来源于云南玉溪,品种为玉溪K326。将烟草取样2.0kg粉碎以95%的甲醇提取 5次,每次提取24h,提取液合并,过滤,减压浓缩成浸膏,得浸膏105g。浸膏用重量2.0倍量的纯甲醇溶解后用120g的100目粗硅胶拌样,0.6kg的160目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,TLC监测合并相同的部分,得到8个部分,其中体积配比为7:3的氯仿-丙酮洗脱部分用安捷仑1100半制备高效液相色谱分离,以36wt%的甲醇为流动相,Zorbax SB-C18(21.2×250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为298nm,每次进样200μL,收集29.5min的色谱峰,多次累加后蒸干;所得产物再次用纯甲醇溶解,再以纯甲醇为流动相,用Sephadex LH-20凝胶柱层析分离,即得该新化合物。
实施例3
烟草样品来源于云南大理,品种为云烟200,将烟草取样3.5kg切碎,以95wt%的乙醇提取4次,每次提取48h,提取液合并,过滤,减压浓缩成浸膏,得浸膏250g。浸膏用重量2.0倍量的纯甲醇溶解后用250g的80目粗硅胶拌样,1.2kg的200目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,TLC监测合并相同的部分,得到8个部分,其中体积配比为7:3的氯仿-丙酮洗脱部分用安捷仑1100半制备高效液相色谱分离,以36wt%的甲醇为流动相,Zorbax SB-C18(21.2×250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为298nm,每次进样200μL,收集29.5min的色谱峰,多次累加后蒸干;所得产物再次用纯甲醇溶解,再以纯甲醇为流动相,用Sephadex LH-20凝胶柱层析分离,即得该新化合物。
实施例4
烟草样品来源于云南昆明,品种为红花大金元,将烟草取样5kg粉碎,以75%的丙酮用超声提取3次,每次提取72h,提取液合并,过滤,减压浓缩成浸膏,得浸膏380g。浸膏用重量比1.6倍量的纯甲醇溶解后用400g的90目粗硅胶拌样,2.4kg的180目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,TLC监测合并相同的部分,得到8个部分,其中体积配比为7:3的氯仿-丙酮洗脱部分用安捷仑1100半制备高效液相色谱分离,以36%的甲醇为流动相,Zorbax SB-C18(21.2×250mm,5μm)制备柱为固定相,流速为20ml/min,紫外检测器检测波长为298nm,每次进 样200μL,收集29.5min的色谱峰,多次累加后蒸干;所得产物再次用纯甲醇溶解,再以纯甲醇为流动相,用Sephadex LH-20凝胶柱层析分离,即得该新化合物。
实施例5
------化合物结构的鉴定
以上述方法制备得到的异吲哚生物碱化合物的结构通过以下方法进行测定。化合物为黄色胶状物,由高分辨质谱可知其准分子离子峰[M+Na]+为304.0790,可推测其分子式为C13H15NO6,化合物的不饱和度为7。紫外光谱在210、260和298nm有吸收峰,说明化合物中有芳环结构。红外光谱在1688,1660,1612,1576,1452cm-1存在强吸收峰,证实化合物中存在羟基、羰基和芳环功能团。根据化合物的1H和13C NMR数据(C-1-C-7a;H2-3,和H-7)(表-1)可初步推测化合物1为异吲哚-1-酮结构。根据H2-3(δH 4.20)和C-1(δC 166.3)、C-3a(δC136.3)、C-4(δC 153.1)、C-7a(δC 116.8);H-7(δH 6.63)和C-1(δC 166.3)、C-3a(δC 136.3)、C-7a(δC 116.8)的HMBC相关(图-3)可进一步证实本发明化合物为异吲哚-1-酮结构。化合物的母核得到确定后,其它基团可归属为取代基,包括一个3-羟基丙酮基(C-1'–C-3',H2-2',H2-3'),一个和氮原子相连的羟乙基(C-1”、C-2”,H2-1”,H2-2”)和两个酚羟基(11.05s、10.84)。根据H2-2'和C-4、C-5和C-6的HMBC相关可证实3-羟基丙酮基取代在苯环的C-5位;羟乙基取代在异吲哚环的氮原子上和由H2-1”和C-1、C-3,以及H2-3和C-1”的HMBC相关确定,根据酚羟基氢(δH 11.05)和C-3a、C-4、C-5,以及(δH10.84)和C-4、C-5、C-6的HMBC相关可证实两个酚羟基取代在C-4和C-6位。至此,本发明化合物的结构的到确定,并命名为4,6-二羟基-2-(2-羟乙基)-5-(3-羟基丙酮基)-异吲哚-1-酮。
实施例6
取实施例3制备的化合物,为黄色胶状物。测定方法与实施例5相同,确认实施例3制备的化合物为所述的异吲哚生物碱化合物——4,6-二羟基-2-(2-羟乙基)-5-(3-羟基丙酮基)-异吲哚-1-酮。
实施例7
取实施例4制备的化合物,为黄色胶状物。测定方法与实施例5相同,确认实施例4制备的化合物为所述的4,6-二羟基-2-(2-羟乙基)-5-(3-羟基丙酮基)-异吲哚-1-酮。
实施例8
取实施例1-4制备的任一异吲哚生物碱化合物进行抗烟草花叶病毒活性试验,试验情况如下:
采用半叶法,在药剂的质量浓度均为50mg/L时对本发明化合物进行抗烟草花叶病毒活性测定。在5~6龄烤烟的植株上,选取适用于测试的叶片(叶行正常,无病无虫),先将叶片均匀撒上细金刚砂,用毛笔将备用的烟草花叶病毒源(3.0×10-3)均匀抹在撒有金刚砂的叶片上,待所有中选的叶片接毒结束后,立即放在盛有药液的培养皿中处理20min,取出,擦去叶片上水珠和药液,将两个半叶复原排放在铺有卫生纸保湿的玻璃缸中,并盖上玻璃盖,控温(23±2)℃,放在温室自然光照射,2~3d即可见枯斑.每个处理都设另一半叶为对照,另外设有1组为商品宁南霉素的处理作为对比,按下公式计算相对抑制率。
XI%=(CK-T)/CK×100%
X:相对抑制率(%),CK:浸泡于清水中半片接毒叶的枯斑数(个),T浸泡于药液中半片接毒叶的枯斑数(个)。
结果明本化合物的相对抑制率为56.8%,超过对照宁南霉素的相对抑制率31.5%,说明化合物有很好的抗烟草花叶病毒活性。
- 还没有人留言评论。精彩留言会获得点赞!