作为盐皮质激素受体拮抗剂的化合物的晶型及其制备方法与流程
本发明属于医药技术领域,具体涉及作为盐皮质激素受体拮抗剂化合物的晶型及其制备方法以及化合物的晶型在制备治疗和/或预防肾损伤或心血管疾病的药物中的应用。
背景技术:
醛固酮是一种在肾上腺皮质合成的盐皮质激素,通过与盐皮质激素受体结合,活化其受体来促进钠离子的保留和钾离子的排泄,对电解质平衡及改变动脉壁上的内皮细胞、血管平滑肌细胞、纤维原细胞和动脉外膜及其介质上的基质的结构和功能具有重要作用。醛固酮水平过高,导致盐皮质激素受体被异常活化,会导致电解质失衡及血管损伤和纤维化等,造成高血压等心血管疾病、肾/心脏/脑等器官损伤以及内分泌紊乱等疾病。药物通过竞争性的与盐皮质激素受体结合,阻断醛固酮与盐皮质激素受体的结合,来抑制醛固酮介导的损害作用,进而减少前述疾病的发生。式(1)所示的化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈(在专利申请WO2012022121中已有描述)为醛固酮受体拮抗剂,选择性与盐皮质激素受体结合,对糖皮质激素、雄激素受体的亲和力较低。WO2012022121记载了式(1)所示化合物的制备方法,是通过式(1)所示化合物的外消旋体化合物拆分后旋干所得,得到的化合物为无定形。式(1)化合物含有两个手性中心,为了得到单一的异构体,本领域技术人员通常需要进行拆分,WO2012022121中记载的是先得到式(1)所示化合物的外消旋化合物后,再进行拆分得到式(1)所示化 合物,使得式(1)所示化合物难以在GMP标准化车间内生产,不利于产业化,且成本较大。
技术实现要素:
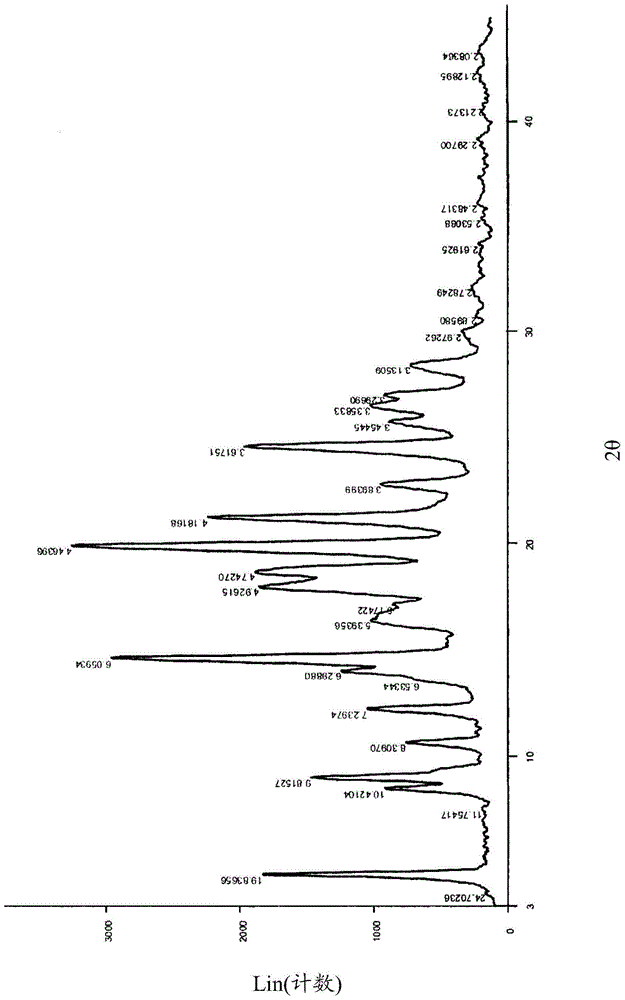
在药物研发中晶型的研究非常重要,化合物的晶型不同,将导致其稳定性、溶解度等性质的不同。因此本发明人对式(1)所示化合物的晶型进行了大量的研究,由此确认和发明了式(1)化合物的晶型。本发明人在制备式(1)化合物时,将拆分步骤提前,使得式(1)所示化合物很容易在GMP标准车间内生产,可以顺利实现产业化。本发明目的之一在于提供式(1)化合物的晶型。本发明目的之二在于提供式(1)化合物的制备方法。本发明目的之三在于提供式(1)化合物的晶型的制备方法及其相互转化方法。本发明的另一目的在于提供式(1)化合物的晶型在预防和/或治疗肾损伤或心血管疾病(包括心脏损伤、高血压、心力衰竭、心肌梗塞、心绞痛、心脏肥大、心肌炎、心脏和血管纤维化、压力感受器官功能障碍或心律不齐)中的应用,及其在制备治疗和/或预防肾损伤或心血管疾病(包括心脏损伤、高血压、心力衰竭、心肌梗塞、心绞痛、心脏肥大、心肌炎、心脏和血管纤维化、压力感受器官功能障碍或心律不齐)的药物中的应用。本发明的技术方案概述如下:1.式(1)所示化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈的晶型其特征在于,使用Cu-Ka辐射,以2θ角度表示的X-射线粉末衍射,在以下位置有特征峰:晶型I:14.8°±0.2°、17.4°±0.2°、19.4°±0.2°、19.8°±0.2°;晶型II:14.6°±0.2°、19.9°±0.2°、21.2°±0.2°、24.6°±0.2°;晶型III:15.3°±0.2°、19.5°±0.2°、20.5°±0.2°、25.0°±0.2°。2.技术方案1所述的化合物的晶型,其特征在于,使用Cu-Ka辐射,以2θ角度表示的X-射线粉末衍射,在以下位置有特征峰:晶型I:14.8°±0.2°、16.9°±0.2°、17.4°±0.2°、19.4°±0.2°、19.8°±0.2°、26.2°±0.2°;晶型II:14.6°±0.2°、18.0°±0.2°、18.7°±0.2°、19.9°±0.2°、21.2°±0.2°、24.6°±0.2°;晶型III:10.0°±0.2°、15.3°±0.2°、15.8°±0.2°、19.5°±0.2°、20.5°±0.2°、25.0°±0.2°。3.技术方案1或2所述的化合物的晶型,其特征在于,使用Cu-Ka辐射,以2θ角度表示的X-射线粉末衍射,在以下位置有特征峰:晶型I:9.8°±0.2°、12.9°±0.2°、14.8°±0.2°、15.4°±0.2°、16.9°±0.2°、17.4°±0.2°、19.4°±0.2°、19.8°±0.2°、22.6°±0.2°、26.2°±0.2°;晶型II:4.5°±0.2°、9.0°±0.2°、12.2°±0.2°、14.0°±0.2°、14.6°±0.2°、18.0°±0.2°、18.7°±0.2°、19.9°±0.2°、21.2°±0.2°、24.6°±0.2°;晶型III:3.8°±0.2°、10.0°±0.2°、15.3°±0.2°、15.8°±0.2°、17.9°±0.2°、19.5°±0.2°、20.5°±0.2°、25.0°±0.2°、26.0°±0.2°、27.2°±0.2°。4.式(1)所示化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈的制备方法,其特征在于,包括以下步骤:(9)将2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯手性拆分得(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯;(10)将(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯水解制得(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸;(11)(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡 唑并[3,4-f]喹啉-7-羧酸与4-羟基哌啶通过缩合反应制得式(1)所示化合物。5.技术方案4的制备方法,还包括下述步骤之一:将步骤(11)得到的式(1)所示化合物置于无水低级醇、乙腈、乙酸乙酯与乙醇的混合溶剂、甲醇与四氢呋喃的混合溶剂、或乙腈与丙酮的混合溶剂中,加热所得到的溶液直至澄清,然后使所述溶液降温,将析出的固体过滤并干燥;或者将步骤(11)得到的式(1)所示化合物置于低级醇中使其溶解,再将所得到的溶液滴入水中,过滤,和任选地真空干燥;或者将步骤(11)得到的式(1)所示化合物用水和乙腈混合溶液浆洗,过滤,和任选地真空干燥;或者将步骤(11)得到的式(1)所示化合物溶于丙酮中,再将所得到的溶液滴入正庚烷中,过滤。5-1.技术方案4或5的制备方法,还包括在步骤(9)前的步骤(8):(8)(E)-6-环戊基亚甲基-5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯与2-氯-4-肼基苯甲腈盐酸盐通过缩合反应制得2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯。5-2.技术方案5-1的制备方法,还包括在步骤(8)前的步骤(7):(7)2-氯-4-肼基苯甲腈与通过盐酸成盐反应制得2-氯-4-肼基苯甲腈盐酸盐。5-3.技术方案5-2的制备方法,还包括在步骤(7)前的步骤(6):(6)2-氯-4-氟苯腈与水合肼通过取代反应制得2-氯-4-肼基苯甲腈。5-4.技术方案5-3的制备方法,还包括在步骤(6)前的步骤(5):(5)5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯与环戊基甲醛通过缩合反应制得(E)-6-环戊基亚甲基-5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯。5-5.技术方案5-4的制备方法,还包括在步骤(5)前的步骤(4):(4)5-氧代-5,6,7,8-四氢喹啉-2-甲腈通过水解和酯化反应得到5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯。5-6.技术方案5-5的制备方法,还包括在步骤(4)前的步骤(3):(3)5-氧代-5,6,7,8-四氢喹啉-N-氧化物与N,N-二甲氨基甲酰氯,三甲基氰硅烷通过取代反应得到5-氧代-5,6,7,8-四氢喹啉-2-甲腈。5-7.技术方案5-6的制备方法,还包括在步骤(3)前的步骤(2):(2)5-氧代-5,6,7,8-四氢喹啉通过氧化反应得到5-氧代-5,6,7,8-四氢喹啉-N-氧化物。5-8.技术方案5-7的制备方法,还包括在步骤(2)前的步骤(1):(1)1,3-环己二酮与乙酸铵,丙烯醛通过缩合和加成反应得到5-氧代-5,6,7,8-四氢喹啉。6.如技术方案1、2或3所述的式(1)所示化合物的晶型I的制备方法,其特征在于,将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈置于无水低级醇、乙腈、乙酸乙酯与乙醇的混合溶剂、甲醇与四氢呋喃的混合溶剂、或乙腈与丙酮的混合溶剂中,加热所得到的溶液直至澄清,然后使所述溶液降温,将析出的固体过滤并干燥得到化合物的晶型I;或者将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈溶于丙酮中,再将所得到的溶液滴入正庚烷中,过滤得到晶型I。7.如技术方案1、2或3所述的式(1)所示化合物的晶型III的制备方法,其特征在于,将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈置于低级醇中使其溶解,再将所得到的溶液滴入水中,过滤得到晶型III;或者将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈用水和乙腈混合溶液浆洗,过滤得到晶型III。8.如技术方案1、2或3所述的式(1)所示化合物的晶型II的制备方法,其特征在于,将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈置于低级醇中使其溶解,再将所得到的溶液滴入水中,过滤得到晶型III;或者将化合物2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈用水和乙腈混合溶液浆洗,过滤得到晶型III;然后再将晶型III真空干燥得晶型II。在本发明中,所述“低级醇”为甲醇、乙醇、正丙醇等。9.一种药物组合物,其特征在于,所述药物组合物含有技术方案1、2或3所述的式(1)所示化合物的晶型,和药用可接受的载体,其中 所述晶型包括晶型I、II、III或其组合。9-1.本发明还提供一种包括式(1)所示化合物的晶型I、II、III或其组合的药物组合物。所述药物组合物还可以包括药用可接受的载体,例如赋形剂、粘合剂、增湿剂、崩解剂、增稠剂等。10.技术方案1、2或3所述的式(1)所示化合物的晶型在制备治疗和/或预防肾损伤或心血管疾病的药物中的应用,其中所述晶型包括晶型I、II、III或其组合。11.技术方案10的应用,其中所述心血管疾病包括心脏损伤、高血压、心力衰竭、心肌梗塞、心绞痛、心脏肥大、心肌炎、心脏和血管纤维化、压力感受器官功能障碍或心律不齐。12.一种治疗和/或预防肾损伤或心血管疾病的方法,所述方法包括给予需要其的受试者治疗有效量的技术方案1、2或3所述的式(1)所示化合物的晶型,其中所述晶型包括晶型I、II、III或其组合。13.技术方案12的方法,其中所述心血管疾病包括心脏损伤、高血压、心力衰竭、心肌梗塞、心绞痛、心脏肥大、心肌炎、心脏和血管纤维化、压力感受器官功能障碍或心律不齐。14.用于治疗和/或预防肾损伤或心血管疾病的技术方案1、2或3所述的式(1)所示化合物的晶型,其中所述晶型包括晶型I、II、III或其组合。15.技术方案14的应用,其中所述心血管疾病包括心脏损伤、高血压、心力衰竭、心肌梗塞、心绞痛、心脏肥大、心肌炎、心脏和血管纤维化、压力感受器官功能障碍或心律不齐。16.式(1)所示化合物的晶型I、II、III及无定形在一定条件下可以相互转化,本发明还提供了晶型I、晶型II、晶型III以及与无定形之间的转化关系。无定形在无水乙醇中重结晶制得晶型I;晶型I、II、III或其组合在低级醇溶剂中溶解后旋干制得无定形;晶型II在无水乙醇中重结晶制得晶型I;无定形溶于甲醇,然后将溶液滴入水中制得晶型III;晶型III室温干燥制得晶型II;晶型I在乙腈与水的体系中浆洗制得晶型III;晶型III在无水乙醇中重结晶制得晶型I。附图说明图1:式(1)化合物晶型I的XRD图谱;图2:式(1)化合物晶型II的XRD图谱;图3:式(1)化合物晶型III的XRD图谱;图4:式(1)化合物晶型I、晶型II、晶型III以及与无定形之间的转化关系图,其中:1.乙醇重结晶;2.低级醇溶解后旋干;3.溶于甲醇,滴入水析出;4.低级醇溶解后旋干;5.低级醇溶解后旋干;6.乙醇重结晶;7.乙腈/水浆洗;8.乙醇重结晶;9.室温干燥。具体实施方式以下通过实施例形式的具体实施方式,对本发明的上述内容作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下实施例。实施例12-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈的制备1、5-氧代-5,6,7,8-四氢喹啉(01)的制备反应方程式:投2个平行反应:在100L的反应釜中,加入45L甲苯,搅拌下加入1,3-环己二酮15kg,加热至固体溶解,加入乙酸铵24kg,加热至回流,反应12h,冷却至0℃,分批缓慢加入丙烯醛共15kg,缓慢升温至回流,反应12h,冷却,分层,下层用甲苯洗涤2次(5L×2),合并甲苯与上层溶液,旋蒸,共得5-氧代-5,6,7,8-四氢喹啉粗品7.4kg,黑色液体,产率18.8%。2、5-氧代-5,6,7,8-四氢喹啉-N-氧化物(02)的制备反应方程式:实验操作如下:将5-氧代-5,6,7,8-四氢喹啉粗品7.4kg投入100L的反应釜中,补加二氯甲烷至总量为50L,降温至-10℃,分批加入间氯过氧苯甲酸共13kg,加入完毕,室温搅拌反应20h,抽滤,滤饼用 二氯甲烷洗涤2次,与滤液合并,用饱和硫代硫酸钠溶液洗涤至碘化钾淀粉试纸不显蓝色,有机相用无水硫酸钠干燥,得溶液50L,不经处理直接投入下一步反应。3、5-氧代-5,6,7,8-四氢喹啉-2-甲腈(03)的制备反应方程式:在100L的反应釜中,加入上步得到的5-氧代-5,6,7,8-四氢喹啉-N-氧化物溶液50L,加入三甲基氰硅烷10kg,缓慢加入N,N-二甲氨基甲酰氯11kg,室温搅拌反应48h,搅拌下缓慢分批加入饱和氢氧化钠水溶液调节pH至8~9,分层,萃取,水相用二氯甲烷洗涤3次(8L×3),合并有机相,用水洗涤1次(20L),有机相用无水硫酸钠干燥,过滤,旋蒸,得约8L红黑色液体,冷却,乙醇结晶,得5-氧代-5,6,7,8-四氢喹啉-2-甲腈930g,产率10.7%(以5-氧代-5,6,7,8-四氢喹啉为原料计算)。4、5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯(05)的制备反应方程式:投3个平行反应:在2L的圆底烧瓶中,加入800mL无水乙醇和5-氧代-5,6,7,8-四氢喹啉-2-甲腈280g,冰浴下加入浓盐酸400mL,升温回流反应16h,冷却,旋蒸,加入1L无水乙醇,冷却至0℃,滴加氯化亚砜200mL,升温至回流反应10h,旋蒸,粗品加入二氯甲烷溶解,用碳酸氢钠水溶液调节pH至碱性,分层,水相用二氯甲烷萃取3次,合并有机相,干燥,旋蒸,共得5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯875g,产率8.18%。5、(E)-6-环戊基亚甲基-5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯(07) 的制备反应方程式:投3个平行反应:在2L的单口瓶中,加入5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯291g和450mL乙醇,-20℃冰浴中加入环戊基甲醛213mL,搅拌10min,缓慢加入吡咯烷110mL,N2置换3次,室温避光反应8h,-20℃冰浴中静置2h,抽滤,固体用冰乙醇洗涤,干燥,共得(E)-6-环戊基亚甲基-5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯862g,产率72.3%。6、2-氯-4-肼基苯甲腈盐酸盐(08)的制备反应方程式:在100L的反应釜中,加入40L乙醇和2-氯-4氟苯腈7kg,加入水合肼4L,加热回流反应5h,冷却,离心,固体投入到100L的反应釜中,加入40L无水乙醇,缓慢加入浓盐酸7.5L,加热回流反应2h,离心,干燥,得2-氯-4-肼基苯甲腈盐酸盐7kg,收率76.2%。7、2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯(09)的制备反应方程式:投4个平行反应:在2L的单口瓶中,加入(E)-6-环戊基亚甲基-5-氧代-5,6,7,8-四氢喹啉-2-羧酸乙酯215.5g、2-氯-4-肼基苯甲腈盐酸盐191g和900mL乙醇,N2置换3次,80℃避光回流反应9h,冷却至室温,-20℃冰浴中静置2h,抽滤,固体用冰乙醇、乙醚分别洗涤,干燥,共得2-(3- 氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯1026g,产率79.3%。8、(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯(12)的制备反应方程式:将2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯用SFC(超临界流体色谱仪)拆分,有两个光学异构体,分离收集第一个组分为(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯;拆分条件:仪器:SFC(Novasep30-50)制备柱:ChiralpakIA,20μm,5×25cm流动相:A相为超临界CO2,B相为二氯甲烷∶四氢呋喃∶二乙醇胺=50∶50∶0.1(vol∶vol∶vol),A∶B=50∶50(vol∶vol)流速150g/min检测波长:465nm样品配置:取2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯1025g,添加二氯甲烷超声溶解,滤过,得浓度约为50mg/mL的样品溶液。用SFC拆分,收集第一个出峰异构体样品即为(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯601.58g。9、(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸(13)的制备反应方程式:在20L的反应釜中,加入(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸乙酯1066g、4L四氢呋喃和2L甲醇,-10℃搅拌10min,缓慢加入溶有氢氧化钠192g的水溶液1.2L,室温反应4h,旋去约一半溶剂,用稀盐酸调pH至3~4,抽滤,固体用冰甲醇和乙醚分别洗涤,干燥,得(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸867g,产率86.7%。10、2-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈(式(1)化合物)的制备反应方程式:在5L的反应瓶中,加入(3S,3aR)-2-(3-氯-4-氰基苯基)-3-环戊基-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-7-羧酸380g、900mL二氯甲烷和360mLN,N-二甲基甲酰胺,搅拌下加入三乙胺380mL,降温至-10℃,继续搅拌10min,加入溶有4-羟基哌啶137g的二氯甲烷溶液700mL,搅拌5min,加入2-(7-偶氮苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯(HATU)380g,室温反应3h,补加2-(7-偶氮苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯50g,反应1h,旋蒸除去二氯甲烷和部分三乙胺,将剩余溶液滴入10倍量水中,有固体析出,抽滤,固体用2L二氯甲烷溶解,用2L水洗涤1次,水相用500mL二氯甲烷反萃1次,合并有机相,干燥,旋干得式(1)固体290g,产率63.7%。分子式:C28H30ClN5O2质谱(M+H):5041H-NMR(CDCl3,400MHz):δ8.375-8.395(1H,d),7.423-7.474(2H,m),7.264-7.276(1H,d),6.968-6.995(1H,dd),4.641-4.678(1H,dd),4.17-4.21(1H,m),3.99(1H,s),3.76-3.79(1H,m),3.515(1H,m), 3.41-3.44(1H,m),3.230-3.322(2H,m),2.995(1H,m),2.321-2.352(1H,m),2.10-2.15(2H,m),1.978-2.089(2H,m),1.863-1.895(1H,m),1.758-1.777(1H,m),1.433-1.663(7H,m),1.221-1.352(2H,m)。实施例22-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(1)将实施例1制备所得式(1)所示化合物1g,置于3mL无水乙醇中,加热至80℃,直至溶液澄清,然后缓慢降温至室温,有大量固体析出,过滤,用无水乙醇淋洗三遍,将得到的固体放置于真空干燥箱中60℃真空干燥12h,获得晶型I。晶型I的X-射线粉末衍射(XRD)图谱如图1中所示,其中主要参数如下:实施例32-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(2)将实施例1制备所得式(1)所示化合物146mg,80℃溶于50mL乙腈中,后缓慢降温至室温,搅拌过夜,过滤,得晶型I。实施例42-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢 -2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(3)将实施例1制备所得式(1)所示化合物200mg,置于100mL圆底烧瓶中,加10mL乙酸乙酯,78℃回流仍不溶解,后加入0.5mL乙醇后80℃搅拌,迅速溶解,缓慢降温至室温,2天后过滤,得晶型I。实施例52-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(4)将实施例1制备所得式(1)所示化合物100mg,置于100mL圆底烧瓶中,加3mL丙酮,溶解,滴入20mL正庚烷中,析出固体,过滤,得晶型I。实施例62-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(5)将实施例1制备所得式(1)所示化合物100mg,置于100mL圆底烧瓶中,取1mL甲醇与1mL四氢呋喃制成甲醇∶四氢呋喃=1∶1的混合溶剂,取0.5mL混合溶剂加入到圆底烧瓶中,60℃加热搅拌溶解,后缓慢降至室温,析出固体,过滤,得晶型I。实施例72-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型I的制备(6)将实施例1制备所得式(1)所示化合物100mg,置于100mL圆底烧瓶中,取5mL乙腈与5mL丙酮配制成乙腈∶丙酮=1∶1的混合溶剂,取3.5mL混合溶剂加入到圆底烧瓶中,60℃加热搅拌溶解,后缓慢降至室温,析出固体,过滤,得晶型I。实施例82-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型III的制备(1)将实施例1制备所得式(1)所示化合物200mg置于4mL甲醇中, 80℃溶解,将该溶液反滴入40mL水中,过滤得到晶型III。晶型III的X-射线粉末衍射(XRD)图谱如图3中所示,其中主要参数如下:实施例92-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型III的制备(2)将实施例1制备所得式(1)所示化合物100mg置于10mL离心管中,取10mL水和1mL乙腈配制成水∶乙腈=10∶1的混合溶液,取8mL水与乙腈的混合溶液加入离心管中,搅拌浆洗,过滤得到晶型III。实施例102-氯-4-[(3S,3aR)-3-环戊基-7-(4-羟基哌啶-1-羰基)-3,3a,4,5-四氢-2H-吡唑并[3,4-f]喹啉-2-基]苯甲腈晶型II的制备将实施例8制备的晶型III室温下真空干燥12h获得晶型II。晶型II的X-射线粉末衍射(XRD)图谱如图2中所示,其中主要参数如下:实验例1本发明化合物晶型I的稳定性供试品:式(1)所示化合物晶型I:通过实施例2制得;影响因素试验考察条件:高温试验:①将式(1)所示化合物晶型I平铺于干燥洁净表面皿中,在60℃ 条件下放置10天,分别于第5天和第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较;②将式(1)所示化合物晶型I内层用药用低密度聚乙烯袋、外套聚酯/铝/聚乙烯药品包装用复合膜密封包装,在60℃条件下放置10天,分别于第5天和第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。高湿试验:将式(1)所示化合物晶型I平铺于干燥洁净表面皿中,在25℃、RH90%±5%条件下放置10天,分别于第5天和第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。光照试验:将式(1)所示化合物晶型I平铺于干燥洁净表面皿中,置光照箱内,于照度为4500Lx±500Lx条件下放置10天,分别于第5天和第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。含量:按照中国药典2010版附录VD高效液相色谱法,采用外标法进行测定。有关物质:按照中国药典2010版附录VD高效液相色谱法,采用面积归一化法进行测定。试验结果:见下表1。表1式(1)所示化合物晶型I的影响因素试验考察结果本发明人对式(1)所示化合物的晶型I的稳定性进行了考察,由试验结果可知,式(1)所示化合物的晶型I在高温、高湿和光照的条件下,有关物质、式(1)所示化合物的含量基本没有变化,晶型I的 稳定性优于无定形,表明式(1)所示化合物的晶型I具有较高的稳定性,便于药品的制备、储存和运输,更利于保证药物使用的有效性和安全性。实验例2本发明化合物晶型II的稳定性供试品:式(1)所示化合物晶型II:通过实施例4制得;影响因素试验考察条件:高温试验:①将式(1)所示化合物晶型II平铺于干燥洁净表面皿中,在60℃条件下放置10天,于第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较;②将式(1)所示化合物晶型II内层用药用低密度聚乙烯袋、外套聚酯/铝/聚乙烯药品包装用复合膜密封包装,在60℃条件下放置10天,于第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。高湿试验:将式(1)所示化合物晶型II平铺于干燥洁净表面皿中,在25℃、RH90%±5%条件下放置10天,于第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。光照试验:将式(1)所示化合物晶型II平铺于干燥洁净表面皿中,置光照箱内,于照度为5000Lx±500Lx条件下放置10天,于第10天取样,测定有关物质及式(1)所示化合物的含量,与0天的样品进行比较。含量:按照中国药典2010版附录VD高效液相色谱法,采用外标法进行测定。有关物质:按照中国药典2010版附录VD高效液相色谱法,采用面积归一化法进行测定。试验结果:见下表2。表2式(1)所示化合物晶型II的影响因素试验考察结果本发明人对式(1)所示化合物的晶型II的稳定性进行了考察,由试验结果可知,式(1)所示化合物的晶型II在高温、高湿和光照的条件下,有关物质、式(1)所示化合物的含量基本没有变化,晶型II的稳定性优于无定形,表明式(1)所示化合物的晶型II具有较高的稳定性,便于药品的制备、储存和运输,更利于保证药物使用的有效性和安全性。实验例3本发明化合物晶型I对高盐诱导的Dahl盐敏感(Dahl/ss)大鼠的器官保护和降压作用供试品及配制:本发明化合物晶型I的固体分散体(本发明化合物晶型I∶PVPK30=1∶8(w/w)):本发明化合物晶型I的固体分散体用适量的灭菌注射用水配制浓度为0.03、0.10、0.30、1.00mg/mL的混悬液,每天临用前配制。动物分组及造模:实验动物:雄性8-9周龄的SPF级(无特定病原体级)Dahl/ss大鼠(购自HARLANLABORATORIES,INC.,由北京维通利华试验动物有限公司代购),正常检疫1周后,体征状况良好的入选本实验。Dahl/ss大鼠根据给药前检测血压随机分为6组:正常对照组(n=10),模型组(4%NaCl,n=12),本发明化合物晶型I治疗组,4组:0.3mg/kg/day剂量组(n=11)、1mg/kg/day剂量组(n=11)、3mg/kg/day剂量组(n=11)、10mg/kg/day剂量组(n=11),n为大鼠只数。实验方法:采用含4%NaCl的AIN-93G试验动物饲料诱导Dahl/ss大鼠高血压肾病模型评价本发明化合物晶型I的体内药效活性。在实验开始前1周,采用夹尾法血压检测方法进行两次血压监测,使大鼠适应血压监测操作。实验开始前监测血压一次,作为实验给药前的基础血压值。大鼠根据给药前检测血压随机分组。第二天开始造模,将含4%NaCl的AIN-93G试验动物饲料用做饲料,动物自由进食和饮水,持续42天。正常对照组给予AIN-93G饲料,模型组及本发明化合物晶型I治疗组的各剂量组给予含4%NaCl的AIN-93G饲料。本发明化合物晶型I治疗组分别给予本发明化合物晶型I0.3、1、3、10mg/kg/day,每天灌胃给药两次,每次灌胃体积为5mL/kg。模型组与正常对照组给予等量的灭菌注射用水。血压(收缩压,简称SBP)检测:每周测量1次血压,连续6周,分析各组血压变化情况。肾脏、心脏病理检测:试验结束后,无痛方式处死大鼠,摘取双侧肾脏、心脏。肾脏用苏木精-伊红(HE)染色,评价肾脏损伤。心脏测量心脏左心室壁厚度,评价心脏损伤。实验结果:降压作用:由此看出,本发明化合物晶型I在该模型上表现出明显的降压作用,并呈现一定的量效关系,见表3。表3SBP(mmHg,平均值±标准差)*p<0.05,与正常对照组相比;#p<0.05,与模型组相比。肾脏和心脏保护:根据肾损伤评分结果,本发明化合物晶型I治疗组在保护肾脏损伤方面有显著的疗效,见表4。与模型组比较,本发明化合物晶型I显著降低左心室壁厚度,见表4。表4对肾脏损和心脏损伤的保护作用*p<0.05,与正常对照组相比;#p<0.05,与模型组相比。在高盐诱导的Dhal/ss大鼠高血压及肾病模型中,本发明化合物晶型I表现出明显的降压作用和肾损伤保护的作用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 盐皮质激素受体拮抗剂及使用方法
- 具有糖皮质激素受体结合活性且具有导入了磺酸酯或磺酰胺结构的苯基作为取代基的新 ...的制作方法
- 具有糖皮质激素受体结合活性的新型1,2-二氢喹啉衍生物的制作方法
- 用于治疗痴呆的糖皮质激素受体拮抗剂的制作方法
- 氯喹或其衍生物与糖皮质激素受体的配体联合应用方法
- 具有糖皮质激素受体结合活性的1
- 用于治疗抑郁症的包含选择性5-羟色胺再摄取抑制剂和糖皮质激素受体拮抗剂的药物组合的制作方法
- 作为黑色素浓集激素受体-1拮抗剂的羟基取代的噻吩并嘧啶酮的制作方法
- 具有取代苯基氨基低级烷基和导入酯结构的苯基作为取代基的新型1,2-二氢喹啉衍生物的制作方法
- 糖皮质激素受体拮抗剂用于治疗感染性病症的用途的制作方法