一种化合物及其制备方法和应用与流程
本发明涉及一种化合物,具体为化合物2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯及其制备方法,及2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯在制备化合物福司氟康唑中的应用。
背景技术:
福司氟康唑是氟康唑的磷酸酯前体药物,有良好的水溶性,可减少输液量,减轻循环系统负担。静脉给药后,在体内被碱性磷酸酯酶水解成氟康唑和磷酸。因此寻找安全、高效的抗真菌感染药物在临床治疗上具有重要的意义。
福司氟康唑(Fosfluconazole),化学名为:2,4-二氟-αα-二(1氢-1,2,4-三唑-1-基甲基)苯甲醇磷酸二氢酯,是对念珠菌属和隐球菌属所致的真菌血症、呼吸器官真菌症、真菌性腹膜炎、消化管真菌症、尿路真菌症、真菌性髓膜炎等有良好疗效。福司氟康唑结构式为:
福司氟康唑由英国辉端公司中央研究所开发,2003年10月16日在日本批准上市,目前文献报道福司氟康唑的合成方法有以下几种:专利WO1997028169公开报道了以氟康唑为原料与四唑、二苄基二异丙基胺基膦发生酯化反应制得氟康唑磷酸酯,与钯碳加氢来脱除苄基制得福司氟康唑,如路线一所示。路线一:
此路线所用原料四唑和二苄基二异丙基胺基膦的价格昂贵、供应量较少,难以进行工业 化生产;最后一步是在甲醇溶剂中通过钯碳催化加氢来脱除苄基,由于福司氟康唑在甲醇中不溶,因此路线有如下的不足之处:催化剂中毒、终产物容易析出、设备操作要求较高、具有安全隐患等,不适合工业化生产。
专利WO1997028169公开报道了以氟康唑为原料与三氯化磷、苯甲醇和过氧化氢发生酯化反应制得氟康唑磷酸酯,与钯碳加氢来脱除苄基制得福司氟康唑,如路线二所示。路线二:
路线二首先采用三氯化磷、苯甲醇和过氧化氢为原料,价格便宜,供应量充足,虽然文献产率为63.0%,反应可控性较差,但可以进行工业化生产,该路线仍然采用钯碳加氢来脱除苄基,不同之处在于把溶剂改为氢氧化钠水溶液,产物在氢氧化钠水溶液中可溶且稳定,但在氢化过程中,原料氟康唑磷酸酯不溶解于氢氧化钠水溶液,导致反应效率较低,反应时间延长,同时容易导致氟康唑磷酸酯水解为氟康唑,使得产品质量难以控制。
专利CN1262551中公布路线如下:
此路线中由路线中的式2化合物采用专利WO1997028169路线二方法制备,经钯碳和甲 酸铵加氢脱除苄基,成铵盐经酸化,制得福司氟康唑,在氢化脱除过程中,由于甲酸铵易升华,很容易堵塞回流冷凝管,导致反应釜过程中压力增加,危险性较高,使得难以进行工业化生产。因此路线有如下的不足之处:催化剂中毒、终产物容易析出、安全隐患较大等,不适合工业化生产。
上述诸技术方案虽各自有其优点,但存在反应可控性较差,反应条件苛刻及危险性较高,成本较高,不利于工业化生产的缺陷。
技术实现要素:
本发明的目的是克服上述现有技术中起始原料价格昂贵,对设备要求苛刻,不易工业化等缺点,提供一种反应步骤简短、反应条件温和、操作简便、对环境友好的福司氟康唑的合成方法。
本发明所指的如式(2)、式(3)以发明内容中指定为准,上述背景技术中涉及的相关式只是为了更好地说明该技术的背景情况,本发明内容中涉及的相关式与上述背景技术中所述指定的有不同,如对于CN1262551中所述的式(2)与本发明所述的式(2)化合物不同。
根据本发明的反应式,本发明提供了一种式(2)的化合物:
式(2)
其中R为叔丁基。
式(2)的化合物作为中间体在制备式(3)化合物福司氟康唑中的应用,式(3)
本发明的式(2)化合物的制备方法,将式(1)化合物氟康唑与三氯化磷、叔丁醇和过氧化氢发生酯化反应制备,
式(1)
本发明的式(2)化合物的制备方法,为将式(1)化合物氟康唑和有机碱加至有机溶剂中,加入三氯化磷使反应,继续加入叔丁醇使反应,加入过氧化氢溶液,使反应,除去水相溶液,用焦亚硫酸钠溶液和水洗涤有机相,蒸去溶剂,并溶剂析晶,过滤,干燥,即得;所述的有机碱选自吡啶、3-甲基吡啶、2,6-二甲基吡啶,有机溶剂选自醚类、酯类、卤代烷类,醚类溶剂可选自四氢呋喃、甲基四氢呋喃,酯类溶剂可选自乙酸乙酯、甲酸乙酯、乙酸丁酯,卤代烷类溶剂可选自二氯甲烷、氯仿、1,2-二氯乙烷、1,1-二氯乙烷。
本发明的式(2)化合物的制备方法,加入三氯化磷使反应和继续加入叔丁醇使反应的反应温度控制为-15~30℃,反应时间可以为0.5~24小时。
本发明的式(2)化合物的制备方法,所述式(1)化合物氟康唑与有机碱的摩尔用量比为1∶(1.0~5.0);所述式(1)化合物氟康唑与三氯化磷的摩尔用量比为1∶(1.0~2.0)。
本发明的式(2)的化合物作为中间体在制备式(3)化合物福司氟康唑中的应用,制备方法为式(2)的化合物在有机溶剂中发生水解反应得到式(3)化合物;水解反应所用酸可以为无机酸,如可选自盐酸、盐酸、硫酸、硝酸、磷酸。
本发明的式(2)的化合物作为中间体在制备式(3)化合物福司氟康唑中的应用,制备方法中有机溶剂选自所用溶剂优选为醇类、酯类或醚类,其中醇类可选自乙醇、甲醇、异丙醇、正丁醇,醚类溶剂可选自四氢呋喃、甲基四氢呋喃、二氧六环,酯类溶剂可选自乙酸乙酯、乙酸甲酯、乙酸丁酯。
本发明的式(2)的化合物制备及作为中间体发生水解得到化合物福司氟康唑反应方程式举例如下:
本发明给出的合成福司氟康唑的新工艺,有益效果为本发明的操作过程无需更低温及无水、无氧等苛刻条件,有效的提高了反应收率,本发明中各步骤反应中所使用试剂均为小分子碱,易于纯化,因此,本发明福司氟康唑的制备方法具有反应条件温和、操作简便、对环境友好、生产成本低等优点,适合于工业化生产。
具体实施方式
通过下述实施例更具体的阐述本发明。然而,所述实施例并不意味着限制本发明的范围。
实施例1
2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯[式(2)]的合成:将氟康唑(765g,2.5mol)和吡啶(593.2g,7.5mol)加到搅拌的乙酸乙酯(3431g)中,然后冷至-5~0℃,滴加三氯化磷(357.1,2.62mol)到反应混合物中,并维持反应温度为-5~0℃反应1小时,于-5~0℃下将叔丁醇(518.0g,7.0mol)滴加入其中,加毕,于-5~0℃下搅拌反应2小时;然后滴加过氧化氢(27.5%w/w水溶液,932.5g),并维持同样温度,于-5~0℃下搅拌反应12小时,除去水相,用焦亚硫酸钠溶液、稀盐酸和水洗涤有机相,减压蒸除溶剂,置于乙酸乙酯(765g)和石油醚(625g)中,在20℃搅拌析晶1小时,在0℃搅拌析晶1小时后,过滤,用乙酸乙酯(76.5g)和石油醚(62.5g)洗涤,然后于50℃真空干燥18小时,得到目标产物(896.8g,72.0%)。
实施例2
2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯[式(2)]的合成:将氟康唑(306g,1.0mol)和吡啶(237.3g,3.0mol)加到搅拌的二氯甲烷(1789g)中,然后冷至-5~0℃,滴加三氯化磷(144.2,1.05mol)到反应混合物中,并维持反应温度为-5~0℃反应1小时,于-5~0℃下将叔丁醇(207.2g,2.8mol)滴加入其中,加毕,于-5~0℃下搅拌反应2小时;然后滴加过氧化氢(27.5%w/w水溶液,373g),并维持同样温度,于-5~0℃下搅拌反应12小时,除去水相,用焦亚硫酸钠溶液、稀盐酸和水洗涤有机相,减压蒸除溶剂,得 到目标产物(333.3g,66.9%)。
实施例3
2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯[式(2)]的合成:将氟康唑(306g,1.0mol)和3-甲基吡啶(279.4g,3.0mol)加到搅拌的乙酸乙酯(1372g)中,然后冷至-5~0℃,滴加三氯化磷(144.2,1.05mol)到反应混合物中,并维持反应温度为-5~0℃反应1小时,控温-5~0℃下将叔丁醇(207.2g,2.8mol)滴加入其中,加毕,于-5~0℃下搅拌反应2小时;然后滴加过氧化氢(27.5%w/w水溶液,373g),并维持同样温度,于-5~0℃下搅拌反应12小时,除去水相,用焦亚硫酸钠溶液、稀盐酸和水洗涤有机相,减压蒸除溶剂,置于乙酸乙酯(306g)和石油醚(250g)中。在20℃搅拌析晶1小时,在0℃搅拌析晶1小时后,过滤,用乙酸乙酯(30.6g)和石油醚(25g)洗涤,然后在50℃真空干燥18小时,得到目标产物(343.3g,68.9%)。
实施例4
福司氟康唑的合成:向反应瓶中,加入2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯(820g,1.64mol),乙醇(4100g),滴加浓盐酸(332.6g,3.28mol),加毕,加热升温至回流,搅拌反应2小时。反应毕,减压蒸出溶剂,加入纯化水(1640g),在15~30℃下搅拌反应2小时,抽滤,无水乙醇(150g)洗涤3次,然后在50℃真空干燥18小时,得到目标产物(580.9g,91.7%)。熔点223~224℃,经结构鉴定与文献一致。
实施例5
福司氟康唑的合成:向反应瓶中,加入2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯(160.0g,0.32mol),二氧六环(800g),滴加浓盐酸(65.1g,0.64mol),加毕,加热升温至回流,搅拌反应2小时。反应毕,减压蒸出溶剂,加入纯化水(300g),在15~30℃下搅拌反应2小时,抽滤,无水乙醇(50g)洗涤3次,然后在50℃真空干燥18小时,得到目标产物(115.9g,93.8%)。熔点223~224℃,经结构鉴定与文献一致。
实施例6
福司氟康唑的合成:向反应瓶中,加入2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基磷酸二叔丁基酯(160.0g,0.32mol),乙酸乙酯(1.2L),滴加30%硫酸(88.2g,0.32mol),加毕,加热升温至回流,搅拌反应2小时。反应毕,减压蒸出溶剂,加入纯化水(300g),在15~30℃下搅拌反应2小时,抽滤,无水乙醇(50g)洗涤3次,然后在50℃真空干燥18小时,得到目标产物(111.2g,90.0%)。熔点223~224℃,经结构鉴定与文献一致。
实施例7
将本发明的式(2)化合物2-(2,4-二氟苯基)-1,3-双(1H-1,2,4-三唑-1-基)-2-丙基 磷酸二叔丁基酯进行结构分析,具体见图1、图2和图3,解析如下:
1H-NMR(500MHz,DMSO)δ=1.29ppm(9H,6×-CH3)、5.02~5.05ppm(2H,-CH2-)、5.13~5.16ppm(2H,-CH2-)、6.95~6.98ppm(1H,-CH=)、7.24~7.29ppm(2H,-CH=)、7.89ppm(2H,2×-CH=)、8.53~8.54ppm(2H,2×-CH=)。
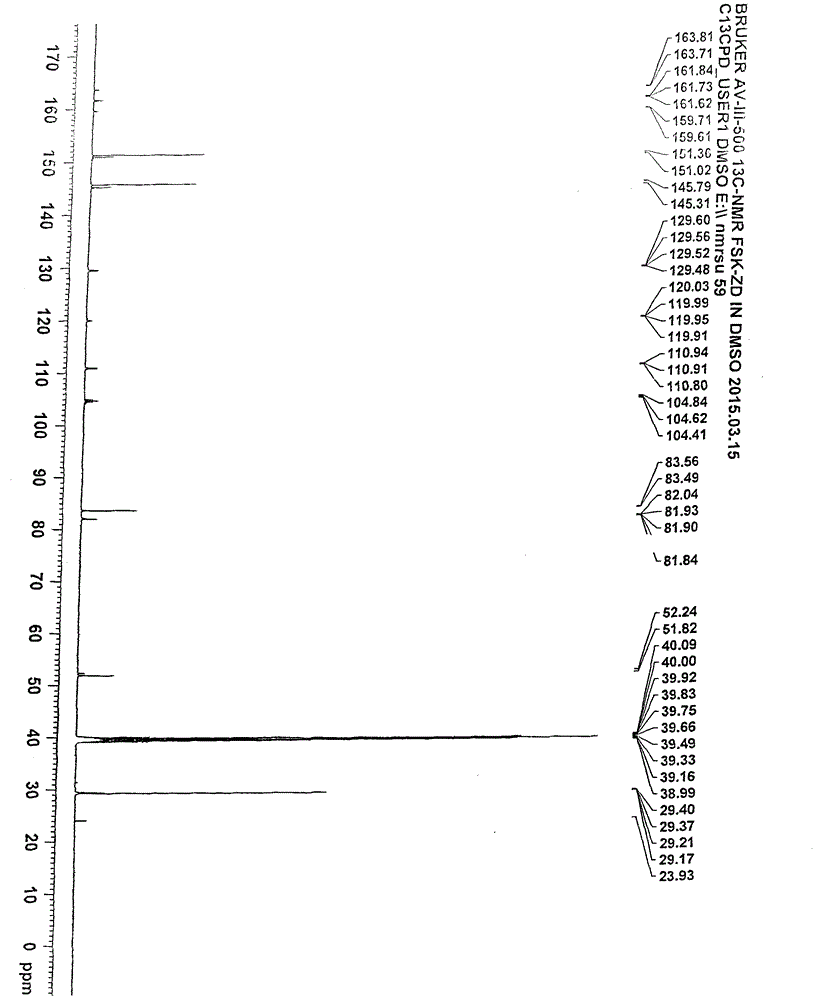
13C-NMR(500MHz,DMSO)δ=23.93ppm(2C,季碳)、29.17~29.40ppm(6C,伯碳)、51.82~52.24ppm(2C,仲碳)、83.49~83.56ppm(1C,季碳)、104.41~104.84ppm(1C,叔碳)、110.80~110.94ppm(1C,叔碳)、119.91~120.03ppm(1C,季碳)、129.48~129.60ppm(1C,叔碳)、145.31~145.79ppm(2C,叔碳)、151.02~151.36ppm(2C,叔碳)、159.61~159.71ppm(1C,季碳)、161.62~161.84ppm(1C,季碳)。
MS:521.2(M+Na+),443.1[M-C4H9+H]+,387.1[M-2C4H9+H]+。
附图说明
图1是本发明实施例式(2)化合物1H-NMR图谱。
图2是本发明实施例式(2)化合物13C-NMR图谱。
图3是本发明实施例式(2)化合物MS图谱。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种他达拉非化合物、及其组合物的制作方法
- 一种新化合物1-[2-(2,4-二甲基苯基硫基)苯基]-2-氧哌嗪及其制备方法和在沃替西汀 ...的制作方法
- 一种抗癌化合物及其用图
- 一种化合物及其制备方法
- 一种叔醇类化合物及其制备方法
- 一种离子射流灯的制作方法
- 一种Mg<sup>2+</sup>,Al<sup>3+</sup>,Zr<sup>4+</sup>,F<sup>-</sup>离子共掺杂石榴石型固体电解质的制作方法
- 一种Mg<sup>2+</sup>,Al<sup>3+</sup>,Zr<sup>4+</sup>,S<sup>2-</sup>离子共掺杂石榴石型固体电解质的制作方法
- 一种热电化合物及其制备方法
- 一种适用于化合物结晶的试管的制作方法