一种4‑芳基香豆素类化合物的应用的制作方法
本发明涉及与内分泌疾病相关的药物技术领域,具体提供一种4-芳基香豆素类化合物的应用。
背景技术:
糖尿病是世界上最常见的代谢紊乱疾病之一,糖尿病在成人中的发病率在过去几十年一直在增加。随着人民生活水平的提高、人口的老龄化和生活方式的改变,糖尿病目前已是继心血管疾病和肿瘤之后的第三大非传染性疾病,给社会和经济带来了沉重的负担,严重威胁人类的健康。糖尿病本身并不可怕,可怕的是它的并发症。糖尿病并发症涉及全身各个器官,是导致血脑血管疾病、肾病、失明、足坏死等严重疾病的主要原因。
葡萄糖苷酶是生物体内糖代谢途径中的重要成员之一。α-葡萄糖苷酶抑制剂是一类以延缓肠道碳水化合物吸收而达到治疗糖尿病的口服降糖药物。α-葡萄糖苷酶抑制剂是比较成熟的治疗糖尿病药物,已广泛应用于临床。其作用机制为:竞争性抑制位于小肠的各种α-葡萄糖苷酶,使淀粉类分解为葡萄糖的速度减慢,从而减缓肠道内葡萄糖的吸收,降低餐后高血糖。α-葡萄糖苷酶抑制剂不刺激β细胞分泌胰岛素,但可降低餐后胰岛素水平,说明可增加胰岛素的敏感性。目前多种α-葡萄糖苷酶抑制剂已经被批准在临床用于糖尿病的治疗,如拜唐苹(阿卡波糖),倍欣(伏格列波糖)。
醛糖还原酶在哺乳动物体内催化葡萄糖向山梨醇的转化,这是糖尿病后遗症如白内障和神经疾病的主要起因。醛糖还原酶抑制剂可有效抑制糖尿病病人许多器官中山梨醇含量的异常升高。而且,能够直接抑制活性氧或氧化应激的抗氧化剂,也是应对糖尿病并发症的重要候选物。
糖基化终产物(advancedglycationendproducts,age)在未患糖尿病受试者组织中缓慢增加,但在糖尿病患者体内,由于循环中持续高糖水平加速了age的产生,使其大量蓄积。产生的过量age不仅可以与蛋白质交联,影响蛋白质性能,也可以通过与特异受体结合,发生反应来改变细胞功能,从而导致机体的病理变化。age与糖尿病肾病、视网膜病变、神经病变、动脉粥样硬化等糖尿病并发症的发生发展密切相关。美国糖尿病控制与并发症实验(dcct)和英国糖尿病前瞻性研究(ukpds)等结果证明,皮肤中age浓度的异常升高是提示糖尿病和未来可能发生并发症的生物标志。
4-芳基香豆素类天然化合物拥有多种药理活性,如抗糖尿病、抗氧化、抗肿瘤、抗疟原虫、抗菌、抗真菌、抗凝血等,具有重要的研究价值和应用潜力。早在20世纪初,就有研究者发现墨西哥药材h.latiflora具有较好的抗糖尿病活性,后来korec课题组发现大鼠口服该类药材的提取物可以起到快速降糖的效果。2005-2007年,该课题组从此类药材中分离纯化出多个4-芳基香豆素的糖苷类化合物,并验证了它们良好的降糖活性,其中化合物i(5-o-β-d-glucopyranosyl-7,3′,4′-trihydroxy-4-phenylcoumarin)活性最好,其结构见下式。
(phytochemistry,2007,68:2087)
前期,本申请人已基于此化合物,合成了多个4-芳基香豆素类化合物(参见cn201610495985.9)。但是,其在抗癌方面的应用上有待提升。
技术实现要素:
本发明的技术任务是针对上述现有技术的不足,提供4-芳基香豆素类化合物的应用。
本发明进一步的技术任务是提供一种预防和/或治疗糖尿病及其并发症的组合物。
为实现上述目的,本发明提供了如下技术方案:
结构式为(ⅰ)的4-芳基香豆素类化合物的应用,
其中,r1、r2、r3、r4为-h、-oh或-och3;
r5、r6、r7为-h、-oh或-och3。
结构式为(ⅰ)的4-芳基香豆素类化合物选自下列化合物:
作为优选,所述化合物用于制备抗氧化剂。
所述抗氧化剂用于清除羟基自由基和dpph自由基活性。
作为优选,所述化合物用于制备α-葡萄糖苷酶抑制剂。
作为优选,所述化合物用于制备ages形成抑制剂。
作为优选,所述化合物用于制备醛糖还原酶抑制剂。
一种抗氧化剂,其有效成分为式(ⅰ)所述4-芳基香豆素类化合物。
一种α-葡萄糖苷酶抑制剂,其有效成分为式(ⅰ)所述4-芳基香豆素类化合物。
一种ages形成抑制剂,其有效成分为式(ⅰ)所述4-芳基香豆素类化合物。
一种醛糖还原酶抑制剂,其有效成分为式(ⅰ)所述4-芳基香豆素类化合物。
申请人发现,以结构式为(ⅰ)的4-芳基香豆素类化合物作为有效成分的抗氧化剂、葡萄糖苷酶抑制剂、ages形成抑制剂、醛糖还原酶抑制剂在预防和/或治疗糖尿病及其并发症方面均具有显著效果。
本发明的预防和/或治疗糖尿病及其并发症的组合物,其有效成分为式(ⅰ)所述4-芳基香豆素类化合物,需要的时候可以加入一种或多种药学上可以接受的载体。所述载体包括药学领域常规的稀释剂、赋形剂、填充剂、粘合剂、润湿剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂等。
所述预防和/或治疗糖尿病及其并发症的组合物为药品或保健品。
本发明所述的预防和/或治疗糖尿病及其并发症的组合物可以经口服或非口服等形式使用,如可通过注射、喷射、滴鼻、滴眼、渗透、吸收、物理或化学介导的方法导入机体如肌肉、皮内、皮下、静脉、粘膜组织;或是被其他物质混合或包裹后导入机体。
用于口服时,可以将其制成常规的固体制剂,如片剂、粉剂、颗粒剂、胶囊、膏剂、霜剂等;制成液体制剂如水或油悬浮剂或其它液体制剂,如口服液等。用于非口服时,可将其制成注射液等。
所述组合物的使用量因使用途径不同而各有不同,对成人来说,作为药品使用时,每天2mg-10mg比较合适;作为保健品使用时,每天2mg-10mg比较合适。
本发明制剂的各种剂型可以按照药学领域的常规生产方法制备。例如将活性成份与一种或多种载体混合,然后将其制成所需剂型。
附图说明
图1.化合物12和13长期降低糖尿病小鼠血糖值的折线图;
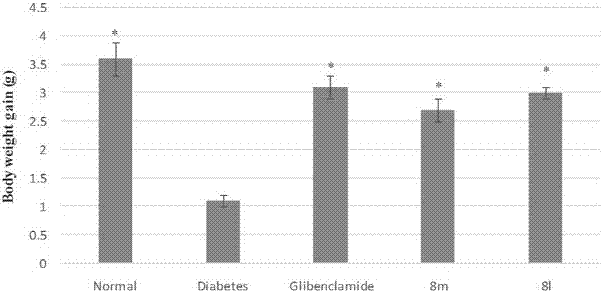
图2.各组小鼠灌胃相应药物16天后的体重变化图;
图3.化合物12和13对糖尿病小鼠糖耐量的影响。
具体实施方式
下面将结合附图和实施例,对本发明的4-芳基香豆素类化合物的应用作进一步详细说明。
下述实施例中所述试验方法,如无特殊说明,均为常规方法;所述试剂和生物材料,如无特殊说明,均可从商业途径获得。
实施例中所用的14种化合物的结构式及编号如下:
【实施例1】14种化合物清除羟基(·oh)自由基和dpph自由基能力的测定
1、清除羟基自由基(·oh)能力的测定方法
h2o2与fe2+反应产生·oh,在体系中加入水杨酸捕捉并产生有色物质,该物质在510nm下有最大吸收。分别取0.3%h2o2,1.0mmo1/lfes04(用10mmol/lh2so4溶液配置),1.0mmo1/l水杨酸(用无水乙醇配置)1.0ml于10.0ml试管中,再加入100-2000μg/ml的梯度浓度的样品溶液各1.0ml。最后以1ml0.3%h2o2启动反应,在37℃水浴中反应30min,以蒸馏水作空白对照,vc做阳性对照,在510nm下测定反应液的吸光值,重复三次试验。
计算羟基自由基的清除率%=[a0-(a1-a2)]/a0×100%,并由此得到ic50值。
式中,a0:1.0ml蒸馏水+1.0mlh2o2+1.0mlfeso4+1.0ml水杨酸+1.0mlh2o2的吸光值;
a1:1.0ml新黄酮类化合物或vc+1.0mlh2o2+1.0mlfeso4+1.0ml水杨酸+1.0mlh2o2的吸光值;
a2:1.0ml新黄酮类化合物或vc+1.0mlh2o2+1.0mlfeso4+1.0ml无水乙醇+1.0mlh2o2的吸光值。
ic50:清除率达到50%时,所需抗氧化剂的浓度(mg/ml)。
2、清除dpph自由基能力的测定方法
准确称取10mgdpph于50ml容量瓶中,用无水乙醇溶解定容,避光保存,使用前稀释成0.04mg/ml使用。取2.0ml浓度为1-1000μg/ml浓度的样品溶液于10.0ml试管中,加入2.0ml上述的dpph溶液,摇匀,避光室温保存30min,用紫外-可见分光光度计于517nm测吸光值,以无水乙醇作空白,vc做阳性对照,重复三次试验。
计算dpph自由基的清除率%=[a0-(a1-a2)]/a0×100%,并由此得到ic50值。
式中,a0:2.0mldpph溶液+2.0ml无水乙醇的吸光值;
a1:2.0mldpph溶液+2.0ml新黄酮类化合物或vc的吸光值;
a2:2.0ml新黄酮类化合物或vc+2.0ml无水乙醇的吸光值。
ic50:清除率达到50%时,所需抗氧化剂的浓度(mg/ml)。
3、试验结果
ic50(halfmaximalinhibitoryconcentration)是指被测量的拮抗剂的半抑制浓度。它能指示某一药物或者物质(抑制剂)在抑制某些生物程序(或者是包含在此程序中的某些物质,比如酶,细胞受体或是微生物)的半量。在自由基清除方面,一定浓度的药物可以清除自由基的生成,使之下降50%,该浓度称为50%抑制浓度。ic50值可以用来衡量药物抗氧化作用的强弱,即抗氧化作用越强,该数值越低。
结果见表1
表114种化合物及vc对羟基自由基(·oh)、dpph自由基的ic50值
4、试验结论
所合成的新黄酮类化合物均从清除2,2-二苯基-1-苦基肼(dpph)自由基和羟基自由基两方面评价其抗氧化活性。实验中使用vc作为阳性对照化合物。大多数化合物表现出中等至优良的清除活性,其中具有骈二羟基取代基的化合物9、11-14显示出优异的活性,强于阳性对照品vc。这些化合物虽然具有相同的母核,但是其苯环上取代基的不同对其抗氧化性能力影响较大。羟基取代基的数量和位置与dpph自由基清除活性相关。通常,羟基取代基的增加和甲氧基取代基的减少可增强dpph自由基清除活性。在清除羟基自由基活性方面,化合物11-14的ic50值为289±0至417±21μg/ml,为vc的2至3倍(ic50=837±24μg/ml),说明具有骈二羟基结构的化合物清除羟基自由基活性较好。
【实施例2】14种化合物体外α-葡萄糖苷酶抑制活性的测定
1、反应溶液的配制
(1)配制底物α-葡萄糖甘(pnpg)溶液:精确称取0.3766gpnpg,加适量0.1mol/l磷酸缓冲液(ph为6.8)溶解,再用容量瓶准确定容到50ml,配制成25mmol/l的母液。将母液分别稀释成0.1、0.5、1.0、2.0、3.0、4.0、5.0mmol/l7个不同梯度的标准品溶液,备用。
(2)配制α-葡萄糖苷酶的酶溶液:将冻干酶粉用0.01mol/l磷酸缓冲液(ph为6.8)溶解,配制成2u/ml的母液。再将酶液分别稀释,配制成0.1、0.2、0.3、0.4、0.5、1.0u/ml的酶溶液,备用。
(3)配制阿卡波糖标准溶液(抑制剂):精确称取10mg阿卡波糖水合物标准品,用容量瓶准确定容到10ml,配制成1000μg/ml的阿卡波糖标准母液。将母液分别稀释成0.05、0.1、0.5、1、5、10、20、40、60μg/ml不同梯度的标准品溶液,备用。
(4)14种化合物均配置成0.1-1000μg/ml浓度梯度的溶液。
(5)0.2mol/l的na2co3:称取2.16gna2co3于烧杯中,加入适量蒸馏水溶解,并定容到100ml,4℃下保存,备用。
2、α-葡萄糖苷酶抑制活性的测定原理
由于pnpg在α-葡萄糖苷酶的作用下能水解产生葡萄糖和pnp,pnp在405nm处有最大吸收,所以测定其吸光度,根据公式便可计算出各样品α-葡萄糖苷酶的抑制率及ic50值。
3、α-葡萄糖苷酶抑制活性的测定方法
测定方法参照masaohattori等试验条件,并做调整。实验分为空白组、对照组、样品空白组和样品组,各反应物按表2中剂量在96孔板中进行加样,每组3个平行。按表2依次加入pbs缓冲液即磷酸缓冲液、测试样品和底物,混合均匀,于37℃水浴保温10min,结束后,取出,加入37℃水浴的酶溶液,充分混匀,于37℃水浴反应20min,结束后加入70μl0.2mol/l的na2co3溶液中止反应。由于pnpg在α-葡萄糖苷酶的作用下能水解产生葡萄糖和pnp,pnp在405nm处有最大吸收,测定其吸光度。
表2各反应物添加计量和顺序(单位:μl)
4、试验结果
根据公式计算出各样品及阿卡波糖对α-葡萄糖苷酶的抑制率及ic50值。
其中,ac为对照组吸光值;ab为空白组吸光值;as为样品组吸光值;asb为样品空白组吸光值。
ic50(halfmaximalinhibitoryconcentration)是指被测量的拮抗剂的半抑制浓度。它能指示某一药物或者物质(抑制剂)在抑制某些生物程序(或者是包含在此程序中的某些物质,比如酶,细胞受体或是微生物)的半量。在酶活力抑制方面,一定浓度的药物可以抑制α-葡萄糖苷酶活力,使之下降50%,该浓度称为50%抑制浓度,即该酶此时的活性是其原有活性的一半。ic50值可以用来衡量药物抑制该酶作用的强弱,即抑制作用越强,该数值越低。
结果见表3。
表314种化合物及阿卡波糖对α-葡萄糖苷酶的ic50值
5、试验结论
尽管所有化合物测试了α-葡萄糖苷酶活性,只有两种化合物6、8、11-14表现出中等至良好的α-葡萄糖苷酶抑制活性。值得注意的是,12(ic50=0.250±0.042μg/ml)具有相对强的活性,稍弱于阿卡波糖(ic50=0.032±0.002μg/ml)。通过比较ic50值,11>13>14,显示具有相邻7,8-二羟基结构比具有5,7-二羟基和6,8二羟基结构的新黄酮类化合物活性弱。化合物8通过脱甲基转化为14后,其ic50值从0.499±0.164μg/ml将至0.400±0.036μg/ml,说明甲氧基取代基不利于抑制α-葡萄糖苷酶。
【实施例3】14种化合物体外ages形成抑制活性的测定
1、ages形成抑制活性测定原理
age具有自发荧光的特性,在近紫外光的照射下,能够在可见光波段发射荧光。在ages的无创荧光快速检测技术研究中,graaff等使用峰值波长为370nm的激发光照射人体皮肤,发现在420~600nm波段产生自体荧光。
2、体外ages形成抑制活性的测定方法
向含有10毫克/ml牛血清白蛋白的50mmpbs(ph7.4)缓冲液中加入0.2m的葡萄糖,并加入0.02%叠氮化钠,以防止细菌增长。此反应混合物(3ml)与各种浓度的目标化合物(1ml)混合。在37℃温育14天后,测定age的荧光强度。用一个荧光分光光度计用激发波长和发射波长分别为350nm和420nm的波长测定。一式三份实验。氨基胍被用作阳性对照化合物。
式中,ac:1.0ml葡萄糖+1.0ml牛血清白蛋白+1.0ml叠氮化钠+1.0mldso的吸光值;
ab:1.0mlpbs+1.0ml牛血清白蛋白+1.0ml叠氮化钠+1.0mldmso的吸光值;
as:1.0ml葡萄糖+1.0ml牛血清白蛋白+1.0ml叠氮化钠+1.0ml待测化合物或氨基胍盐酸盐的吸光值;
asb:1.0mlpbs+1.0ml牛血清白蛋白+1.0ml叠氮化钠+1.0ml待测化合物或氨基胍盐酸盐的吸光值。
3、试验结果
14种化合物及氨基胍盐酸盐对ages形成的ic50值
ic50(halfmaximalinhibitoryconcentration)是指被测量的拮抗剂的半抑制浓度。它能指示某一药物或者物质(抑制剂)在抑制某些生物程序(或者是包含在此程序中的某些物质,比如酶,细胞受体或是微生物)的半量。在ages形成抑制方面,一定浓度的药物可以抑制ages的形成,ic50值可以用来衡量药物抑制ages形成作用的强弱,即抑制作用越强,该数值越低。
结果见表4。
表414种化合物及氨基胍盐酸盐对ages形成的ic50值
4、试验结论
结果表明大多数4-芳基香豆素类化合对糖基化终产物ages呈现较强的抑制性,甚至优于氨基胍盐酸盐(ic50=31.265±0.942μg/ml)。其中活性最为显著的化合物的ic50值大小顺序为14<11<12<2<3<13<3.0μg/ml,为阳性对照的10倍以上。当所述4-芳基香豆素类化合都具有3',4'-二羟基取代基时,同时具有6,8-二羟基的化合物比具有7,8-二羟基的化合物活性好,并且比具有5,7-二羟基的化合物活性更强。一系列ic50值,11<12<3<2<5,表明具有7,8-二羟基的化合物随着甲氧基取代基的增加,活性减弱。
【实施例4】化合物11、12、13和14四种化合物体外醛糖还原酶抑制活性的测定
综合以上三个实施例中的试验测定结果,选择各项活性均较好的化合物11、12、13和14进行醛糖还原酶的测定。
1、醛糖还原酶(arl2)的提取
醛糖还原酶从sd大鼠晶状体中提取。采用已报道的方法,从大鼠眼球中得到,然后做了部分提纯。虽然没有得到高纯度的醛糖还原酶,但并不影响测试。由于酶在高温时很容易失活,所有提取操作在低于4℃进行。从正常杀死的大鼠眼球中迅速提取出晶状体,然后加入3倍于其体积的冷去离子水(0-4℃),再用匀浆机匀浆。匀浆液在低温离心机中以12000g转速,0-4℃的温度,离心30min。最后取上清液,即为醛糖还原酶的水溶液,用于酶活性测试。
2、体外醛糖还原酶活性测定原理
在nadph的存在下,醛糖还原酶催化dl-甘油醛还原为甘油,同时nadph转化为nadp+,而还原态nadph在340nm处有特征吸收,可以测定340nm处光密度的下降速率△abs,间接测定醛糖还原酶的活性。
3、醛糖还原酶活性的测定
采用秦祥宇的方法进行测定,酶的活性最佳值是处于nadph吸光度变化在0.011±0.0010吸光度单位/min的范围内,如果不在此范围,通过调整酶液浓度来使其达到这一范围。对测试比色皿要加对照比色皿是为了校正由于非酶因素(比如空气中氧气也会氧化nadph)所导致的nadph的氧化。所加试剂顺序及用量见下表,各反应试剂均提前在25℃水浴中预热,混合后20s在340nm波长下,用紫外监测5min,记录下数据,求斜率记为i0。
表5醛糖还原酶酶活性测定各反应物添加量
4、体外醛糖还原酶(alr2)抑制活性的测定
样品管反应体系:按照表6所示,分别加入ph6.2的0.2mol/l磷酸缓冲液1.1ml,0.10mmol/lnadph四钠盐1.25ml,10mmol/ldl-甘油醛1.25ml,0.5ml的醛糖还原酶水溶液,15ul被测化合物溶液,混匀,20s后开始在紫外分光光度计上测定△abs,测定波长340nm,测定时间5min,温度为25℃,求斜率记为ix。对照组测定:样品用dmso溶剂代替;阳性对照组测定:用槲皮素测定。
表6醛糖还原酶抑制测定各反应物添加量
5、试验结果
计算4种化合物对醛糖还原酶的抑制率及ic50值。
ar抑制率计算:不同提取物对醛糖还原酶的抑制作用采用抑制的百分率表示,计算公式如下:抑制率(%)=(∣i0-ix∣/∣i0∣)×100%。
对醛糖还原酶的ic50值:
ic50(halfmaximalinhibitoryconcentration)是指被测量的拮抗剂的半抑制浓度。它能指示某一药物或者物质(抑制剂)在抑制某些生物程序(或者是包含在此程序中的某些物质,比如酶,细胞受体或是微生物)的半量。在酶活力抑制方面,一定浓度的药物可以抑制醛糖还原酶活力,使之下降50%,该浓度称为50%抑制浓度,即该酶此时的活性是其原有活性的一半。ic50值可以用来衡量药物抑制该酶作用的强弱,即抑制作用越强,该数值越低。计算ic50值时,所得出的浓度为被测样品在体系中的最终浓度,而不是样品本身配置的浓度。
结果见表7。
表74种化合物及槲皮素对醛糖还原酶的ic50值
6、试验结论
根据以上已测定实验结果,选取11-14四个化合物进行醛糖还原酶抑制活性的测定。结果见表7,它们的ic50值从小到大为0.203±0.001、0.357±0.015、4.815±0.189、4.97±0.052ug/ml。其alr2抑制的效力是阳性对照药品槲皮素的0.2-5.5倍。其中化合物11和12对alr2表现出最强的抑制活性,优于槲皮素(ic50=1.119±0.088μg/ml)。这表明具有相邻7,8-二羟基的4-芳基香豆素类化合物在抑制alr2方面更有效。
【试验实施例1】化合物12、13和14对小鼠急毒性实验测定
一、试验方法
根据体外活性筛选结果,选取化合物12、13、14三个化合物进行小鼠急毒性实验以及小鼠体内抗糖尿病活性测定。参照myrna的方法,选择昆明小鼠(雄性和雌性各半,体重范围,18-22g)进行实验。将小鼠置于具有12小时明/暗循环的光控室中,实验前12小时禁食,但动物可以自由饮水。将待测化合物用含有0.2%吐温-80的生理盐水溶液配制,调节浓度以0.2ml/10g体重的剂量灌胃给药。实验分为两个阶段进行,每个阶段持续14天,只灌喂一次:第一阶段,分为10,100和1000mg/kg三个剂量组,每组6只小鼠;第二阶段,将剂量调节至1600,2900和5000mg/kg三个剂量组,每组6只小鼠。在两个阶段中,每天观察小鼠的死亡率,毒性作用和/或行为模式的变化。实验结束后处死动物。
二、实验结果与分析
经过两个阶段的实验,灌胃三种药物各个剂量的小鼠均没有死亡,表明化合物12、13、14的半数致死率(ld50)均>5000mg/kg,对人类基本无毒。不需要进行其半数致死率的进一步精确测定。
【试验实施例2】化合物12、13和14对糖尿病小鼠急性抗高血糖试验
一、试验方法
1、stz诱导糖尿病
选用雄性昆明小鼠(18-23g),在昼夜交替恒温(20-250c)控制的环境适应性饲养3天。配置ph为4.5的0.1m柠檬酸缓冲溶液,现配现用,并用该溶液溶解链脉佐菌素(stz)。小鼠禁食不禁水12小时后,以110mg/kgstz的剂量腹腔注射。7天后测量空腹血糖值,将空腹血糖值>11.1mmol/l定为糖尿病小鼠。
2、分组及给药
取66只糖尿病小鼠,按空腹血糖水平随机分成11组,每组6只。
(1)模型组:灌胃给药相应体积的含有0.05%吐温-80的生理盐水;
(2)阳性对照组:灌胃给药10mg/kg的格列本脲,用含有0.05%吐温-80的生理盐水配制;
(3)化合物12分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制;
(4)化合物13分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制;
(5)化合物14分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制。
二、血糖测量及结果分析
1、血糖测量:
血糖测量用拜耳血糖仪测量糖尿病小鼠灌胃药物后0,1.5,3,5,7,9小时的空腹血糖值。
血糖相对于0时的变化百分比采用以下公式计算:血糖变化百分比(%)=[(gi–gt)/gi]·100%
其中,gi为初始(0h)血糖值;gt为小鼠灌胃药物后各个时间点的血糖值。
2、结果分析
结果见表8。
表83种化合物快速降低糖尿病小鼠血糖的变化百分比
每个值都是六只小鼠血糖变化百分比的平均值±标准差。
*在同一时间p<0.05与模型组比较(n=6)。
如表8所示,与模型组相比三个化合物(12、13和14)的三个剂量组(10mg/kg,30mg/kg和100mg/kg)及阳性对照药品格列本脲(10mg/kg)对stz诱导的糖尿病小鼠均具有良好至显著的急性降糖活性。其中,化合物12(30mg/kg)导致血糖水平的降低最显著(p<0.05),灌胃该药物后,糖尿病小鼠血糖降低百分比大于50%,并且持续9个小时(如表8示)。化合物12(30和100mg/kg)在7小时观察到最强的抗高血糖效应(分别为77.14%和70.56%)。剂量为30mg/kg的化合物13和14在9小时对糖尿病小鼠具有最高抗高血糖效应(分别为64.20%和62.77%),整个实验过程中二者显示出相似的抗糖尿病活性,而化合物12稍强于13。阳性对照品格列本脲(10mg/kg)在9小时显示最大低血糖效应(70.81%)。从整个结果中看出化合物12(30mg/kg)在3、5、7小时血糖降低百分比高于格列本脲(10mg/kg),但是在相同剂量时却不如格列本脲。而化合物13和14均低于格列本脲在灌胃后9小时内血糖降低水平。
【试验实施例3】化合物12、13和14对正常小鼠急性降血糖试验
一、试验方法
分组及给药
取66只健康雄性昆明小鼠,按空腹血糖水平随机分成11组,每组6只。
(1)正常组:灌胃给药相应体积的含有0.05%吐温-80的生理盐水;
(2)阳性对照组:灌胃给药10mg/kg的格列本脲,用含有0.05%吐温-80的生理盐水配制;
(3)化合物12分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制;
(4)化合物13分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制;
(5)化合物14分为低、中、高剂量组(10mg/kg、30mg/kg、100mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制。
二、血糖测量及结果分析
1、血糖测量:
用拜耳血糖仪测量健康小鼠灌胃药物后0,1.5,3,5,7,9小时的空腹血糖值。
血糖相对于0时的变化百分比采用以下公式计算:
血糖变化百分比(%)=[(gi–gt)/gi]·100%
其中,gi为初始(0h)血糖值;gt为小鼠灌胃药物后各个时间点的血糖值。
2、结果分析
结果见表9。
表93种化合物快速降低正常小鼠血糖值变化百分比
每个值都是六只小鼠血糖变化百分比的平均值±标准差
*在同一时间p<0.05与正常组比较(n=6)。
在正常小鼠中,化合物12,13和14(10mg/kg,30mg/kg和100mg/kg)的给药组与正常组比较,降低血糖水平不显著,而格列本脲组在1.5、3和5小时与正常组比较明显降低血糖(p<0.05)(如表9示)。
综合试验实施例2和试验实施例3发现化合物12对糖尿病小鼠及正常小鼠均有较强的降糖效果,其中效果最好的浓度为30mg/kg;另外浓度为30mg/kg的化合物13(化合物i的苷元)对糖尿病小鼠降糖活性也较强。所以选择化合物12和13进行糖尿病小鼠长期抗糖尿病活性的测定,灌胃浓度选为30mg/kg。
【试验实施例4】化合物12和13对糖尿病小鼠长期降血糖效果的试验
一、试验方法
1、stz诱导糖尿病
同试验实施例2。
2、分组及给药
取30只糖尿病小鼠,按空腹血糖水平随机分成5组,每组6只。
(1)正常组:灌胃给药相应体积的含有0.05%吐温-80的生理盐水;
(2)模型组:灌胃给药相应体积的含有0.05%吐温-80的生理盐水;
(3)阳性对照组:灌胃给药10mg/kg的格列本脲,用含有0.05%吐温-80的生理盐水配制;
(4)化合物12(30mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制;
(5)化合物13(30mg/kg)灌胃给药,用含有0.05%吐温-80的生理盐水配制。
每天给药1次,早上9:00~10:00,连续给药16天。
二、血糖测量及结果分析
1、血糖测量
糖尿病小鼠长期降糖试验的评价指标为1,4,7,10,13,16天灌胃药物后的空腹血糖值,以及小鼠16天的体重变化。结果分别见图1及图2。
图1中,每个值都是六只小鼠体重增长的平均值±标准差,
*p<0.05与模型组比较(n=6)。
图2中,每个值都是六只小鼠体重增长的平均值±标准差,
*p<0.05与模型组比较(n=6)。
由以上结果可以看出化合物12和化合物13对糖尿病小鼠具有良好的治疗作用,且这两组小鼠与正常组小鼠相比没有明显的消瘦现象。
【试验实施例5】化合物12和13的口服葡萄糖耐量(ogtt)试验
小鼠灌胃药物15天后,禁食不禁水12小时后灌胃相应药物,半小时后,灌胃葡萄糖(2g/kg),测定其0,0.5,1,2,3小时空腹血糖值变化。
血糖相对于0时的变化百分比采用以下公式计算:
血糖变化百分比(%)=[(gi–gt)/gi]·100%
其中,gi为初始(0h)血糖值;gt为小鼠灌胃药物后各个时间点的血糖值。
结果见图3。
图3中,每个值都是六只小鼠血糖变化百分比的平均值±标准差,
*在同一时间p<0.05与模型组比较(n=6)。
结果表明化合物12可以改善糖尿病小鼠糖耐量能力,降低其血糖值。
综合以上所有实施案例可以看出,化合物12具有良好的治疗糖尿病活性,且无毒。通过清除自由基活性、抑制α-葡萄糖苷酶活性、抑制糖基化终产物(ages)的生成、抑制醛糖还原酶活性等多种途径发挥降低糖尿病病人餐后血糖以及治疗糖尿病并发症的作用。有希望发展为多靶点治疗糖尿病的药物,具有重要的研究价值和应用潜力。
以上所述的实施例,只是本发明较优选的具体实施方式,本领域的技术人员在本发明技术方案范围内进行的通常变化和替换都应包含在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!