丙型肝炎病毒辅助受体CD36的特异性抑制剂在制备防治丙型肝炎病毒感染药物中的应用的制作方法
本发明涉及一种丙型肝炎病毒(hcv)辅助受体cd36蛋白的特异性抑制剂在防治hcv感染中的应用,特别涉及hcv辅助受体cd36蛋白的特异性抑制剂—磺基-n-琥珀酰亚胺油酸酯(sulfo-n-succinimidyloleate,sso)在防治hcv感染中的应用,本发明属于生物医药领域。
背景技术:
丙型肝炎是一种由丙型肝炎病毒(hepatitiscvirus,hcv)感染引起的病毒性肝炎,呈全球性流行,据世界卫生组织筛查,过去15年,全球至少有1.85亿人感染hcv,且每年新发丙型肝炎病例约3~4万例。据统计,hcv急性感染后,大约86%的感染者不能自发清除病毒而变为慢性感染,继而发展为慢性肝脏疾病,10%~20%的感染者进一步恶化造成肝硬化,约7%的肝硬化患者发展为肝细胞癌(hepatocellularcarcinoma,hcc),最终导致肝衰竭甚至死亡,每年大约有130万人死于hcv慢性感染及相关疾病。而且,40%~74%的慢性感染患者伴有冷球蛋白血症、周围神经病变、雷诺氏综合征以及干燥综合征等肝外疾病。在未来20年,由hcv感染引起的肝脏疾病所导致的死亡将继续上升。因此,hcv感染被认为是全球性的重大疾病,已成为严重的社会经济和公共卫生问题。据卫生部公布的数据,近年来我国hcv感染报告病例数呈逐年上升的趋势。
hcv存在非常高的基因多态性,目前普遍将hcv病毒株分为7种主要的基因型和67种亚型,各个基因型的全球地理性分布相对复杂,全世界发现最多的是1a和1b,被称为“流行亚型”。我国的感染率为0.43%,中国大陆最常见的基因型是1b和2a,二者流行率为91.6%。
hcv是高复制病毒,在患者体内,每天可产生大约1万亿的病毒颗粒,由于病毒rna多聚酶没有校正功能,复制具有高突变性,长期用药易诱导产生耐药病毒株,能够逃脱宿主的免疫反应而使得hcv疫苗以及泛基因型抗病毒药物的研发面临巨大挑战。到目前为止,依然没有hcv的保护性疫苗能够满足临床的需要。过 去几十年,长效干扰素(peg-ifn-α)联合利巴韦林(ribavirin,rbv)一直是欧洲肝脏研究学会(easl)批准的慢性丙型肝炎患者的标准治疗方案,但感染患者的持续病毒应答率较低,且伴有严重的副作用。近年来国外研发小分子化合物,通过直接作用于病毒自身蛋白来抑制病毒的复制,初步研究显示了很好的临床应用前景,少数几个已批准上市,还有部分直接抗病毒药物(daas)正处于临床在研,有望获批用于hcv感染的治疗,但这些化合物存在诸多缺点,如化合物主要是针对基因1型而其它基因型效果不好、不同程度地出现耐药问题、对以往无效患者的总体疗效明显降低、对难治性患者的不可用药问题、副作用问题使患者的依从性不好影响疗效、靶点有限导致联合方案有限、以及费用昂贵等,这将是今后抗hcv药物研发所要面临的巨大挑战。
hcv依赖宿主存活并完成复制周期,近几年来,宿主蛋白的深入探索为hcv感染的治疗和抗hcv药物的开发提供了新的策略和方向。通过调节宿主环境来抑制病毒的复制,与daas联合治疗慢性丙型肝炎,有望降低病毒的耐药突变率从而达到更好的治疗效果,逐渐成为抗hcv药物研发的热点,也是解决耐药问题和对各种基因型hcv感染都有效的一个新思路。
在以宿主蛋白为靶点的抗hcv药物的研究中,本发明证实,宿主分化簇36(clusterofdifferentiation36,cd36)蛋白的特异性抑制剂磺基-n-琥珀酰亚胺油酸酯(sulfo-n-succinimidyloleate,sso)通过阻断cd36蛋白的辅助受体功能而显示出强效的抗hcv活性。本发明提出了sso的抗hcv作用,迄今为止未见国内外相关文献报道,研发此类药物能够为临床抗hcv药物特别是抗耐药hcv药物的研发提供新的思路和视角。
技术实现要素:
本发明的目的之一是提供丙型肝炎病毒(hcv)辅助受体cd36蛋白的特异性抑制剂,特别是磺基-n-琥珀酰亚胺油酸酯(sulfo-n-succinimidyloleate,sso)在制备防治hcv感染药物中的应用;
本发明的目的之二是提供一种抗hcv的药物组合物。
本发明的目的之三是提供所述药物组合物在制备防治hcv感染药物中的应用。
为了达到上述目的,本发明采用了以下技术手段:
本发明首先检测了上述sso的体内安全性和体外毒性,然后检测了sso的体外抗hcv活性以及与临床已知抗hcv药物的联合用药效果,并分析了所述sso 对耐药hcv的抑制活性,最后分析了sso的作用机制。
本发明通过研究首次发现并证实hcv辅助受体细胞膜表面蛋白cd36的一种特异性抑制剂sso能够通过直接与cd36结合,阻断hcv感染的早期阶段而发挥强效的抗病毒活性,具有很高的体外治疗指数。sso的抗病毒机制不同于目前所有已知的临床和在研的抗病毒药物。sso与已知临床应用的抗病毒药物(vx-950或ifn-α)联合应用,能够显著增强vx-950和ifn-α的抗病毒活性,且证实sso对已知临床用药telaprevir、simeprevir或sofosbuvir引起的耐药病毒株也有显著的抑制作用。另外,实验证实口服sso后安全性好,对小鼠体重、存活率以及肝肾功能没有影响。
因此,在上述研究的基础上,本发明提出了hcv辅助受体cd36蛋白的特异性抑制剂在制备防治hcv感染药物中的应用。
在本发明中,优选的,所述的hcv辅助受体cd36蛋白的特异性抑制剂为磺基-n-琥珀酰亚胺油酸酯(sulfo-n-succinimidyloleate,sso),其结构如式(i)所示:
在本发明中,所述的丙型肝炎病毒为hcv野生型或hcv耐药病毒株。
在本发明中,优选的,所述的hcv耐药病毒株为hcv蛋白酶抑制剂telaprevir耐药株、hcv蛋白酶抑制剂simeprevir耐药株或hcv多聚酶抑制剂sofosbuvir耐药株。
进一步的,本发明还提出了一种抗hcv的药物组合物,其含有磺基-n-琥珀酰亚胺基油酸酯以及另外一种或多种抗hcv的药物。
在本发明,优选的,所述的药物组合物中的另外一种或多种抗hcv的药物包括但不限于特拉匹韦(vx-950)、α-干扰素(ifn-α)或其组合。
在本发明,优选的,所述的药物组合物中还含有一种或多种药学上可接受的载体。
再进一步的,本发明还提出了所述的药物组合物在制备防治hcv感染药物中 的应用。
本发明所述的cd36特异性抑制剂,特别是sso有望开发成为新型的防治hcv感染的药物,并有望在将来通过与已知药物联合用药,来完全清除丙肝病人体内的hcv病毒,尤其是可用于防治hcv感染导致的肝移植患者的hcv再感染。
附图说明
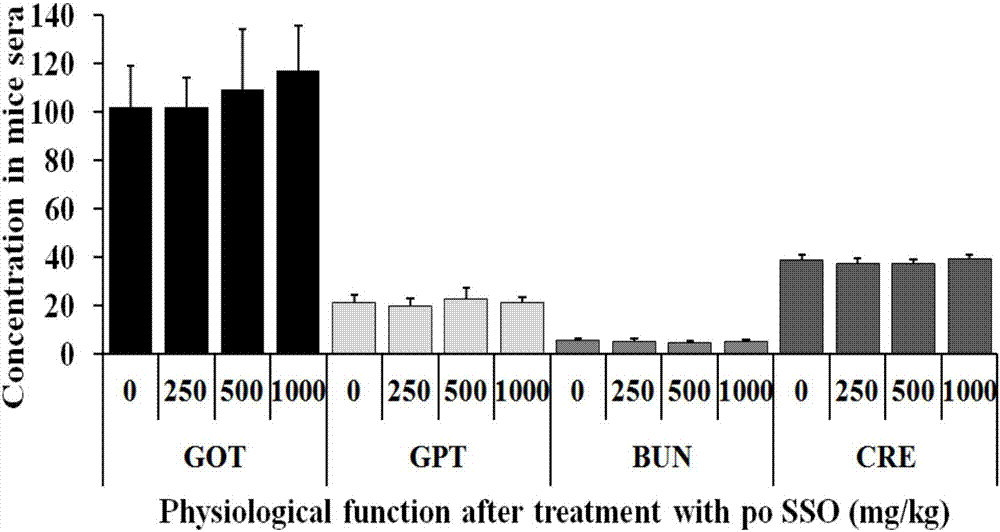
图1为sso对小鼠体重的影响(n=10);
图2为sso对小鼠肝肾功能的影响(n=10);
图3为sso在细胞内抑制hcv复制(n=3);
图3a为sso的细胞毒性以及rna水平抗hcv活性,图3b为蛋白水平抗hcv活性;
图4为sso增强vx-950的体外抗hcv活性;
图4a为rna水平抗hcv活性(n=4);图4b为蛋白水平抗hcv活性(n=4);**,与溶剂对照组比较,p<0.01;#,与vx-950单用组比较,p<0.05;
图5为sso增强ifn-α的体外抗hcv活性;
图5a为rna水平抗hcv活性(n=3);图5b为蛋白水平抗hcv活性(n=3);*,与溶剂对照组比较,p<0.05;**,与溶剂对照组比较,p<0.01;#,与ifn-α单用组比较,p<0.05;
图6为sso抑制hcv生活周期的早期阶段(before,感染前2小时加药处理细胞2小时);
图6a为rna水平抗hcv活性(n=3);图6b为蛋白水平抗hcv活性(n=4);**,与溶剂对照组比较,p<0.01;
图7为sso抑制hcv生活周期的早期阶段(during,感染同时加药处理细胞2小时);
图7a为rna水平抗hcv活性(n=3);图7b为蛋白水平抗hcv活性(n=4);**,与溶剂对照组比较,p<0.01;
图8为sso抑制hcv生活周期的早期阶段(after,感染后2小时加药处理细胞2小时);
图8a为rna水平抗hcv活性(n=3);图8b为蛋白水平抗hcv活性(n=3);
图9为biacore实时分析sso与cd36蛋白间的相互作用曲线;
大图为sso单浓度与cd36蛋白之间相互作用曲线;右下角插入的小图为浓度 梯度的sso与cd36蛋白之间相互作用曲线;
图10为cd36表达水平对sso抗hcv活性的影响。
图10a为cd36低表达增强sso活性(n=3);图10b为cd36高表达减弱sso活性(n=3)。*,与完全空白对照组比较,p<0.05;**,与完全空白对照组比较,p<0.01;##,与sso单用比较,p<0.01。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。这些实施例仅用于例证目的,绝不对本发明的范围构成任何限制。
本发明中体外实验所用sso均用dmso配制成20mm的母液,保存于-20℃,临用前用完全培养液稀释至工作浓度。体内实验中sso用5%tween-80配制成相应浓度。
实施例1体外活性和安全性测定
1.1体内安全性测定
昆明种小鼠,4周龄,40只,体重19.0±1.0g。适应性喂养2天后(约21.0±1.0g),随机分为4组,每组10只,雌雄各半,实验动物使用许可证号为syxk(京)2012–0021。动物分组及给药情况如表1所示,给药前,小鼠称重并禁食4~6小时。然后每组小鼠分别一次性灌胃给予对照溶媒(5%tween-80)或250mg/kg、500mg/kg和1000mg/kgsso并禁食4小时。给药后小鼠正常饲养7天,每天观察小鼠存活率和体重变化,第7天称重并采集血液样本,检测样本中谷草转氨酶(glutamateoxaloacetatetransaminase,got)、谷丙转氨酶(glutamate-pyruvatetransaminase,gpt)、尿素(bloodureanitrogen,bun)和肌酐(creatinine,cre)水平,以分析sso的体内安全性。
表1动物分组及给药情况
结果:
sso对小鼠体重的影响如图1所示,sso对小鼠肝肾功能的影响如图2所示,从上述结果可以看出口服sso后安全性好,对小鼠体重、存活率以及肝肾功能没有影响。
1.2细胞毒性测定
收集生长良好的细胞(huh7.5细胞或gs4.3细胞),用含10%胎牛血清的dmem培养液配细胞悬液,接种于96孔板内,每孔100μl,1×104个细胞/孔,37℃,5%co2孵箱培养24h后,加入以3倍梯度稀释的sso药物溶液,每个浓度设3个平行孔,最高浓度为100μm,并设置等量的dmso作为对照。药物处理72h后,每孔加入mtt溶液10μl(5mg/ml),继续培养4h后吸弃上清液,每孔加100μldmso,震荡10分钟以充分溶解甲瓒结晶。然后以630nm为参考波长,测定570nm处的吸光值。reed&muench法计算半数细胞毒性浓度(halfmaximalcytotoxicconcentration,cc50),结果如表2及图3所示。
表2sso的抗hcv活性
1.3体外抗hcv活性
huh7.5细胞以3×104个/cm2密度种入6孔板或96孔板。培养24小时后,细胞感染hcv-2a(感染量为45iu/细胞)的同时,用不同浓度的sso药物溶液处理,并以等量的dmso作为对照。处理72小时后,提取细胞内总rna(96孔板和6孔板)和总蛋白(6孔板),分别定量hcvrna以及hcvcore和ns3蛋白以分析化合物的体外抗hcv活性,并计算半数有效浓度(halfmaximaleffectiveconcentration,ec50)。
结果如图3及表1所示,图3a为sso的细胞毒性以及rna水平抗hcv活性,图3b为sso蛋白水平抗hcv活性。
以上结果表明,sso对huh7.5细胞没有毒性,且sso能够抑制病毒rna的复制及hcvcore和ns3蛋白的合成,即sso具有体外抗病毒的活性。
实施例2sso增强已知抗hcv药物的活性
huh7.5细胞感染hcv-2a(感染量为45iu/细胞)的同时,用sso(1.0μm)联合vx-950(0.1μm)或联合ifn-α(1.0u/ml)处理,以等量的dmso作为对照。72小时后提取细胞内总rna和总蛋白,分别定量hcvrna以及hcvcore和ns3蛋白以分析sso对vx-950或ifn-α的抗病毒活性的影响。
结果如图4及图5所示,以上结果表明:相较于单独使用,sso与已知临床应用的抗病毒药物(vx-950或ifn-α)联合应用,能够显著增强vx-950和ifn-α的抗病毒活性,说明sso与vx-950或ifn-α之间存在协同作用。
实施例3sso具有抑制耐药hcv感染的活性
通过点突变,构建hcv耐药病毒株(蛋白酶抑制剂telaprevir耐药株(hcv-a156t)、蛋白酶抑制剂simeprevir耐药株(hcv-s282t)和多聚酶抑制剂sofosbuvir耐药株(hcv-d168v)),以检测sso抗耐药病毒的活性。huh7.5细胞以3×104个/cm2密度种入96孔板。培养24小时后,细胞感染耐药hcv(感染量为45iu/细胞)的同时,用不同浓度的sso药物溶液处理,以相应的耐药药物作为阴性对照,以等量的dmso作为空白对照。处理72小时后,提取细胞内总rna,定量hcvrna以分析sso的体外抗耐药hcv活性,并计算ec50,结果如下表3-表5所示。
表3sso抗telaprevir耐药hcv活性
注:wt(wildtype)为野生型hcv病毒株;hcv-a156t为telaprevir耐药病毒株;耐药倍数为药物抗突变株活性ec50和抗野生株ec50的比值。
表4sso抗sofosbuvir耐药hcv活性
注:wt(wildtype)为野生型hcv病毒株;hcv-s282t为sofosbuvir耐药病毒 株;耐药倍数为药物抗突变株活性ec50和抗野生株ec50的比值。
表5sso抗simerevir耐药hcv活性
注:wt(wildtype)为野生型hcv病毒株;hcv-d168v为simeprevir耐药病毒株;耐药倍数为药物抗突变株活性ec50和抗野生株ec50的比值。
以上结果表明,sso对已知临床用药telaprevir、simeprevir或sofosbuvir引起的耐药病毒株也有显著的抑制作用。
实施例4sso治疗hcv感染的作用机制
4.1sso抑制hcv感染的阶段
huh7.5细胞以3×104个/cm2的密度种入6孔板培养。培养24小时后,感染hcv-2a,感染量为150iu/细胞。分别在感染前2小时(before)、感染的同时(during)或感染后2小时(after),各加入5μm、25μm的sso溶液处理,以等量的dmso作溶剂对照。hcv感染或sso处理2小时后,弃去药物或/和感染液,细胞用pbs洗涤1次,加入新的完全培养液继续培养。72小时后,提取细胞内总rna和总蛋白,分别定量hcvrna以及hcvcore和ns3蛋白以分析sso抑制hcv复制的环节。结果如图6-图8所示。
图6a为hcv生活周期的早期阶段(before,感染前加药预处理细胞2小时)中,sso在rna水平的抗hcv活性;图6b为sso在蛋白水平的抗hcv活性。图7a为hcv生活周期的早期阶段(during,感染同时加药处理细胞2小时)中,sso在rna水平的抗hcv活性;图7b为sso在蛋白水平的抗hcv活性,图8a为hcv生活周期的早期阶段(after,感染后加药处理细胞2小时)中,sso在rna水平的抗hcv活性;图8b为sso在蛋白水平的抗hcv活性。
以上结果表明,sso主要通过阻断hcv感染的早期阶段而发挥强效的抗病毒活性。
4.2sso与膜蛋白cd36的相互作用
4.2.1sso与cd36直接结合
采用biacore技术,实时观察小分子化合物sso与cd36之间的相互作用。首先在293t/17细胞中,通过转染高表达质粒cd36-ha诱导蛋白表达,借助融合的ha标签蛋白纯化得到目的蛋白cd36-ha,进行浓缩、脱盐纯化并定量。然后,将蛋白cd36-ha分别用ph4.0、4.5、5.0和5.5的醋酸钠缓冲液稀释成200μl浓度为80μg/ml的溶液,进行预富集,确定最佳偶条件均为ph4.5。接着用ph4.5的醋酸钠缓冲液将cd36-ha稀释至80μg/ml,通过氨基偶联的方式,将其偶联到cm5芯片上,并用nhs和edc活化芯片,用乙醇胺封闭(ethanolamine)芯片。偶联结束后,cd36-ha的响应值分别为4800ru。用浓度为50μm的sso测试结合反应,并用不同的再生液再生芯片。并以单循环的方式,用浓度为0μm、50μm、100μm、200μm和500μm的sso溶液测试结合反应。
图9为采用biacore实时分析sso与cd36蛋白间的相互作用曲线。大图为sso单浓度与cd36蛋白之间相互作用曲线;右下角插入的小图为浓度梯度的sso与cd36蛋白之间相互作用曲线。
以上结果表明:sso通过直接结合膜蛋白cd36发挥抗hcv活性。
4.2.2cd36表达水平影响sso药效
huh7.5细胞以4.5×104个/cm2密度种入6孔板培养。培养24小时后,转染0.2μgpcdna3.1(+)-cd36质粒(以等量的pcdna3.1(+)作对照)或150pmolcd36sirna(以等量的controlsirna-a作对照),转染48h后,感染hcv-2a(感染量为150iu/细胞)的同时用5μm的sso溶液处理。2h后,细胞用pbs漂洗一次,然后加入正常培养液培养。96h后,提取细胞内总rna,定量hcvrna以分析cd36表达水平对sso抗hcv药效的影响。
图10为cd36表达水平对sso抗hcv活性的影响。该结果表明cd36低表达能够增强sso活性(图10a),而cd36高表达能够减弱sso活性(图10b)。
以上结果表明:sso通过直接与cd36结合,阻断hcv感染的早期阶段而发挥强效的抗病毒活性。
以上所述内容仅为本发明的优选实施例,对于本发明而言仅是说明性的,而绝非限制性的。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但都将落入本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 作为AXL抑制剂的杂环化合物的制作方法与工艺
- HPTP‑β抑制剂的制作方法与工艺
- 作为LSD1抑制剂的环丙胺的制作方法与工艺
- KRAS G12C的抑制剂的制作方法与工艺
- 调节雌激素受体突变体的方法和组合物与流程
- 作为PI3K抑制剂的吡啶并[1,2‑A]嘧啶酮类似物的制作方法与工艺
- 作为PI3K抑制剂的吡啶并[1,2‑A]嘧啶酮类似物的制作方法与工艺
- 包含Ⅲ类受体酪氨酸激酶抑制剂和烷基化组蛋白‑脱乙酰酶抑制剂融合分子EDO‑S101的药物组合及其在治疗癌症中的用途的制作方法与工艺
- 包含Ⅲ类受体酪氨酸激酶抑制剂和烷基化组蛋白‑脱乙酰酶抑制剂融合分子EDO‑S101的药物组合及其在治疗癌症中的用途的制作方法与工艺
- 新型雌激素相关受体α抑制剂及其医学用途的制作方法与工艺
- 作为原肌球蛋白受体激酶(Trk)抑制剂的取代的吡唑并[1,5‑a]吡啶的制作方法与工艺
- 一种核孤儿受体TR3抑制剂多肽及其应用的制作方法与工艺
- 程序性死亡受体1基因抑制剂及其制备方法与应用与流程
- 作为H3受体抑制剂的包含哌啶和哌嗪环的氨基甲酸酯/脲衍生物的制作方法与工艺
- 一种雄性激素受体抑制剂的结晶形式及其制备方法与流程
- 一种雄性激素受体抑制剂的结晶形式及其制备方法与流程
- 丙型肝炎病毒辅助受体CD36的特异性抑制剂在制备防治丙型肝炎病毒感染药物中的应用的制造方法与工艺
- IRE‑1α抑制剂的制造方法与工艺
- 白细胞介素‑23受体的口服肽抑制剂以及其治疗炎症性肠病的用途的制造方法与工艺
- 一类孪药型HMG‑CoA还原酶抑制剂及其合成方法与制造工艺
- Flt3蛋白在制备肝癌对索拉菲尼疗效评估试剂盒中的应用
- 用于治疗癌症的雄激素受体抑制剂与akt抑制剂的组合的制作方法
- 一种药物中间体2-芳酰基吲哚衍生物的合成方法
- 一种作用于EGFR敏感突变激酶EGFR<sup>L858R</sup>,EGFR<sup>(d746-750)</sup>的6-取代氨基嘌呤类化合物及 ...的制作方法
- 一种6-溴喹啉的制备方法
- 一种基于巢式复合cold-pcr的t790m突变检测方法
- 具抗肿瘤活性的表皮生长因子受体酪氨酸激酶抑制剂irsf和irsh及其制备方法和应用
- 具抗肿瘤活性的表皮生长因子受体酪氨酸激酶抑制剂irsf和irsh及其制备方法和应用
- 一种多维替尼的制备方法
- 具有抗肿瘤活性的表皮生长因子受体酪氨酸激酶抑制剂nxgf和nxgh及其制备方法和应用