一种恩替卡韦固体分散体和一种恩替卡韦制剂的制作方法
本发明涉及医药药品
技术领域:
,具体涉及一种恩替卡韦固体分散体和一种恩替卡韦制剂。
背景技术:
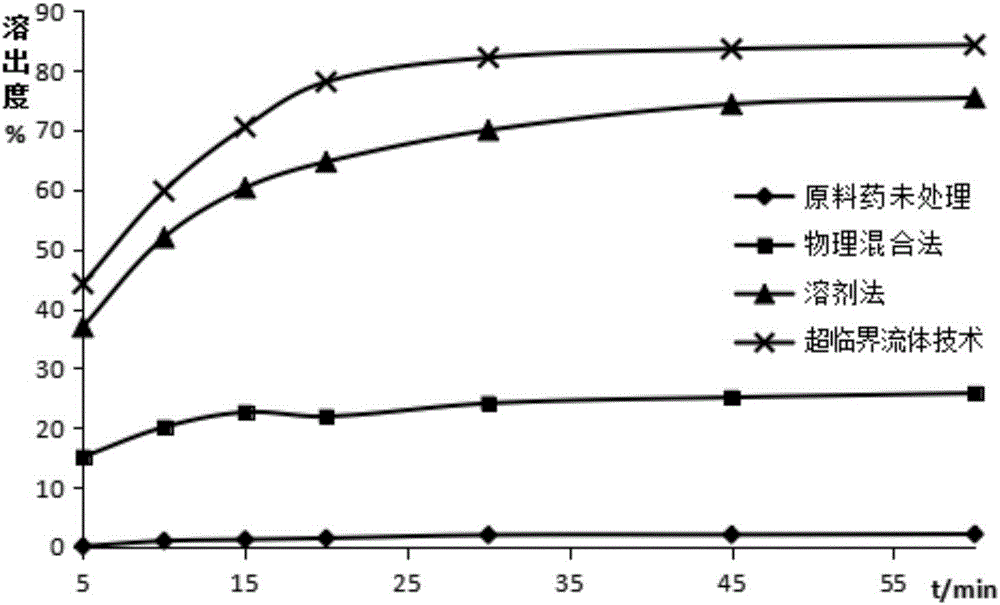
:恩替卡韦是一种高效的抗病毒剂,对乙肝病毒具有良好的抑制作用。分子式为:C12H15N5O3·H2O,分子量为295.3,其结构式为:由于其在制剂中的含量很低,只含有0.5mg或1mg,在制剂过程中主药量与辅料量相差悬殊,不易混匀,故而易造成含量均匀度不合格现象。CN102144983B将原料药与辅料按等量递增的方法进行混合,提高了制剂分散均匀性、含量均匀度,但并未能有效的解决恩替卡韦的溶出度问题。药物的溶解度和分散状态是影响药物体内生物利用度的主要因素,恩替卡韦为一种水难溶性药物,常温状态下,其在水中的溶解度仅为2.4mg/ml,因此其在胃肠道的吸收较低,故而限制了它的临床应用。CN102100677A公开了一种恩替卡韦分散片及其制备方法,该制剂采用包合技术将微粉化的恩替卡韦与β-环糊精混合制成β-环糊精包合物,再与其他辅料混合,制粒,压片而成,但该技术需要超微粉碎设备,同时应用过程中存在批间粒度差异,导致批间疗效差异,且粉碎后,产生大量静电和粉体粒子,静电作用强,对人体危害大,并且微粉粒子容易聚集造成后果是粉体流动性大,难以混合均匀,反而影响药物的溶出。目前,常规的制备恩替卡韦固体分散体的技术主要有溶剂法、熔融法和研磨法等。如CN1046158A公开了一种由喷雾干燥法或溶剂法或冷冻干燥法制得恩替卡韦固体分散体的口腔崩解片的方法;CN102727444A公开了一种将恩替卡韦和乳糖溶解或分散于溶剂中,加热搅拌,去除溶剂,干燥粉碎制备而成的固体分散体;CN101224211A、CN1011812224A采用溶剂法制备固体分散体;CN1020087447A公开了一种将恩替卡韦和吐温-80的溶媒加入到熔融后的载体中制备固体分散体的方法;CN103177017A公开了在恩替卡韦和羟丙基纤维素的乙醇溶液中加入交联聚维酮制备固体分散体的方法。但这些方法都存在一些不足。溶剂法需要使用大量的有机溶剂,环境污染严重,并且有机溶剂残留会引起人体的毒副作用;熔融法加热和冷却速率难以控制,批间重现性差,制备条件剧烈,容易破坏药物的活性;研磨法得到的固体分散体还存在难于粉碎和过筛等问题,也限制了其进一步制备成药物制剂。技术实现要素:本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提供一种恩替卡韦固体分散体和一种恩替卡韦制剂。具体地,本发明提供了一种恩替卡韦固体分散体,其特征在于,所述恩替卡韦固体分散体由恩替卡韦与亲水性载体通过超临界流体方法制备得到。作为本发明的进一步改进,所述恩替卡韦与亲水性载体的质量比为1:1~1:20,优选为1:6。作为本发明的进一步改进,所述亲水性载体选自聚乙二醇类、泊洛沙姆类、纤维素类、糊静、乳糖、甘露醇、硅胶中的一种或多种,优选为聚乙二醇与甘露醇按质量比为4:1的混合物。作为本发明的进一步改进,所述超临界流体方法包括:将恩替卡韦与亲水性载体混合后转入高压反应釜中进行反应,反应压力为20~30MPa,温度为30~60℃,反应完成后粉碎,得到所述恩替卡韦固体分散体。作为本发明的进一步改进,反应压力为25MPa,反应温度为50℃。作为本发明的进一步改进,反应时间为6~24小时,优选为12小时。本发明还提供了一种恩替卡韦制剂,所述恩替卡韦制剂由本发明提供的恩替卡韦固体分散体和药学辅料组成。作为本发明的进一步改进,所述药学辅料包括稀释剂、崩解剂、润滑剂中的一种或多种。作为本发明的进一步改进,所述稀释剂选自乳糖、淀粉、糊精、微晶纤维素中的一种或多种,优选为乳糖与糊精按质量比为1:2的混合物;所述稀释剂的用量为恩替卡韦制剂总质量的60%~90%。作为本发明的进一步改进,所述润滑剂包括苯甲酸钠、醋酸钠、氯化钠、聚氧乙烯单硬脂酸酯、聚氧乙烯月桂醇醚、DL-亮氨酸、月桂醇硫酸钠、月桂醇硫酸镁、聚乙二醇4000或6000中的一种或多种,优选为月桂醇硫酸镁;更优选,月桂醇硫酸镁的用量为恩替卡韦制剂总质量的0.1%-1.0%。作为本发明的进一步改进,所述恩替卡韦制剂的制备步骤包括:S1、先将稀释剂与部分崩解剂混合得到混合辅料;S2、将所述恩替卡韦固体分散体分散于溶剂中形成混合体系,然后该混合体系喷洒于所述混合辅料中,得到软材并制粒,烘干得到整粒;S3、往S2的整粒中再加入剩余崩解剂和润滑剂,混合均匀后成型,得到所述恩替卡韦制剂。作为本发明的进一步改进,所述崩解剂包括内加崩解剂与外加崩解剂,且所述内加崩解剂在步骤S1中使用,所述外加崩解剂在步骤S3中使用;所述内加崩解剂为交联聚维酮(PVPP),外加崩解剂为交联羧甲基纤维素钠(CMC-Na);所述崩解剂的用量为恩替卡韦制剂总质量的10%~20%。作为本发明的进一步改进,步骤S2中,所述溶剂为水、乙醇中的一种或多种,优选为水;溶剂的用量为恩替卡韦固体分散体的质量的1~5倍;烘干温度为55~30℃。作为本发明的进一步改进,所述恩替卡韦制剂为片剂、胶囊、颗粒或口服液,优选为分散片。本发明提供的恩替卡韦固体分散体和恩替卡韦制剂,利用固体分散技术,使药物以分子或无定形状态分散于载体中,可以保证药物的高度分散状态,有效地提高难溶性药物的溶解度和溶出速度,从而提高药物的生物利用度。本发明的附加方面的优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。附图说明本发明的上述和/或附加的方面和优点从结合下面附图对实施方式的描述中将变得明显和容易理解。图1为载体A、B、C、恩替卡韦原料药D和恩替卡韦固体分散体S1、DS1-DS3的DSC曲线图。图2为恩替卡韦原料药D、恩替卡韦固体分散体S1、DS1-DS2的体外溶出曲线。图3为恩替卡韦固体分散体S2-S5的溶出度曲线。图4为恩替卡韦固体分散体S6-S11的溶出度曲线。图5为恩替卡韦固体分散体S12-S16的溶出度曲线。图6为恩替卡韦固体分散体S17-S20的溶出度曲线。图7为恩替卡韦固体分散体S21-S23的溶出度曲线。具体实施方式为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。具体地,本发明提供了一种恩替卡韦固体分散体,所述恩替卡韦固体分散体由恩替卡韦与亲水性载体通过超临界流体方法制备得到。本发明提供的恩替卡韦固体分散体,利用固体分散技术,使药物以分子或无定形状态分散于载体中,可以保证药物的高度分散状态,有效地提高难溶性药物的溶解度和溶出速度,从而提高药物的生物利用度。作为本发明的进一步改进,所述恩替卡韦与亲水性载体的质量比为1:1~1:20,优选为1:6。作为本发明的进一步改进,所述亲水性载体选自聚乙二醇类、泊洛沙姆类、纤维素类、糊精、乳糖、甘露醇、硅胶中的一种或多种。优选情况下,所述亲水性载体为聚乙二醇与甘露醇按质量比为4:1的混合物。本发明的发明人通过大量实验发现,当亲水性载体采用聚乙二醇与甘露醇按质量比为4:1的混合物时,所得到的恩替卡韦固体分散体的溶出度最高。超临界具有溶解好、传质能力高、表面张力低、粘度低及渗透性强等特点,基于超临界流体这些独特的物理化学性质,将超临界流体技术用于制备固体分散体具有工艺流程简单、无溶剂残留(或者溶剂残留相比其他方法最低)、温度低、产品无需干燥以及不影响药物活性等传统方法不可比拟的优势。具体地,本发明中,所述超临界流体方法包括:将恩替卡韦与亲水性载体混合后转入高压反应釜中进行反应,反应压力为20~30MPa,温度为30~60℃,反应完成后粉碎,得到所述恩替卡韦固体分散体。本发明采用超临界流体技术,将恩替卡韦与亲水性载体一起放入反应釜中,通过设定特定的反应压力、温度及时间,制备恩替卡韦固体分散体。经差示量热(DSC)试验显示,超临界流体技术制备的恩替卡韦固体分散体晶体几乎完全消失,这也就意味着恩替卡韦可以无定形状态或分子状态存在于固体分散体中,从而有利于提升药物在水中的溶解性;此外,经溶出度试验结果显示,与常规的溶剂法、物理混合法制备的恩替卡韦固体分散体相比,恩替卡韦的分散状态好,本发明采用的超临界流体技术制备的恩替卡韦固体分散体的溶出度最高,溶出效果最佳。本发明的发明人通过大量实验发现,反应压力为25MPa时得到的恩替卡韦固体分散体的溶出度最高。另外,本发明的发明人通过大量实验发现,反应温度为50℃时得到的恩替卡韦固体分散体的溶出度最高。作为本发明的进一步改进,反应时间为6~24小时。本发明的发明人通过大量实验发现,当反应时间为12h和24h时,固体分散体中药物在60min的累积溶出度相差不多,从节约能耗方面考虑,最终确定反应时间优选为12小时。进一步地,本发明还提供了一种恩替卡韦制剂,所述恩替卡韦制剂由本发明提供的恩替卡韦固体分散体和药学辅料组成。如前所述,本发明提供的恩替卡韦制剂,其中含有恩替卡韦固体分散体,即其利用固体分散技术,使药物以分子或无定形状态分散于载体中,可以保证药物的高度分散状态,有效地提高难溶性药物的溶解度和溶出速度,从而提高药物的生物利用度。其中,所述药学辅料可以采用本领域技术人员常用的各种药学辅料,本发明没有特殊限定。作为本发明的进一步改进,所述药学辅料包括稀释剂、崩解剂、润滑剂中的一种或多种。作为本发明的进一步改进,所述稀释剂选自乳糖、淀粉、糊精、微晶纤维素中的一种或多种,但不局限于此。优选情况下,所述稀释剂为乳糖与糊精按质量比为1:2的混合物。本发明中,所述稀释剂的用量为恩替卡韦制剂总质量的60%~90%。本发明中,稀释剂的主要作用是用来填充制剂的重量或体积,以便于压片。作为本发明的进一步改进,所述润滑剂包括苯甲酸钠、醋酸钠、氯化钠、聚氧乙烯单硬脂酸酯、聚氧乙烯月桂醇醚、DL-亮氨酸、月桂醇硫酸钠、月桂醇硫酸镁、聚乙二醇4000或6000中的一种或多种,但不局限于此。优选情况下,所述润滑剂为月桂醇硫酸镁。更优选情况下,月桂醇硫酸镁的用量为恩替卡韦制剂总质量的0.1%-1.0%。本发明中,所述恩替卡韦制剂可以采用常规方法制备方法,不同之处仅在于其中恩替卡韦原料药采用本发明提供的采用超临界流体技术方法制备得到的恩替卡韦固体分散体。作为本发明的进一步改进,所述恩替卡韦制剂的制备步骤包括:S1、先将稀释剂与部分崩解剂混合得到混合辅料;S2、将所述恩替卡韦固体分散体分散于溶剂中形成混合体系,然后该混合体系喷洒于所述混合辅料中,得到软材并制粒,干燥得到整粒;S3、往S2的整粒中再加入剩余崩解剂和润滑剂,混合均匀后成型,得到所述恩替卡韦制剂。作为本发明的进一步改进,所述崩解剂包括内加崩解剂与外加崩解剂,且所述内加崩解剂在步骤S1中使用,所述外加崩解剂在步骤S3中使用。其中,所述内加崩解剂为交联聚维酮,外加崩解剂为交联羧甲基纤维素钠。本发明中,所述崩解剂的用量为恩替卡韦制剂总质量的10%~20%。作为本发明的进一步改进,步骤S2中,所述溶剂为水、乙醇中的一种或多种,优选为水,但不局限于此。其中,溶剂的用量没有特殊限定,只需要将恩替卡韦固体分散体分散均匀即可。优选情况下,溶剂的用量为恩替卡韦固体分散体的质量的1~5倍。根据本发明的方法,将含有恩替卡韦固体分散体的混合体系喷洒与混合辅料上,制备软材后进行制粒,得到湿颗粒。本发明中,对于湿颗粒的粒径没有较大限制,优选情况下,过24目筛即可。根据本发明的方法,然后对湿颗粒进行干燥处理,得到整粒。其中干燥可采用烘干处理,但不局限于此。优选情况下,烘干温度为55~30℃。烘干后过30目筛,得到整粒。然后往整粒中加入外加崩解剂和润滑剂,混合均匀后成型,即可得到所述恩替卡韦制剂。本发明中,所述恩替卡韦制剂为片剂、胶囊、颗粒或口服液,优选为分散片。恩替卡韦无臭,无味,熔点大于260℃,不具引湿性,性质稳定,可以开发多种剂型满足临床需要。恩替卡韦为水难溶性药物,适合制成固体口服制剂,如片剂、胶囊、颗粒等,不适宜制备注射类剂型。原研厂家为中美上海施贵宝制药有限公司普通片剂和口服液。每片仅含0.5mg恩替卡韦,含量很小。因此采用常规辅料或者普通的工艺无法确保混合均匀或者达到理想溶出效果。本发明中,优选将所述恩替卡韦制剂制备成分散片,由于分散片对生产条件无特殊要求、制造工艺同普通片剂、无需特殊包装、生产成本低和服用方法多样,尤其适合于老人和吞咽困难患者。所述成型的方法为本领域技术人员所公知,例如制备片剂,如分散片,所述成型即为压片。综上,本发明提供的恩替卡韦固体分散体和恩替卡韦制剂,相对于现有技术,具有以下优点。(1)本发明选用一种新型技术——超临界流体技术制备恩替卡韦固体分散体,与传统技术相比,它具有操作条件易于控制,制得微粒的粒径大小、粒度分布以及粒子形态结构的重现性好;处理过程温和,避免了微粒在常规制粒过程中产生相转变、高表面能静电和化学降解等缺点;绿色环保,无污染,降低了药物制剂的生产成本优势。(2)难溶性药物恩替卡韦在固体分散体中以无定型状态存在,与恩替卡韦原粉相比,所制备的恩替卡韦固体分散体流动性好、不粘连、粒径小、分布均匀、性质稳定,体外溶出速率明显改善,生物利用度显著提高。(3)参考国内外上市普通片剂有关情况,本发明制备的恩替卡韦分散片的规格确定为0.5mg,片重在70mg~90mg之间。显然,主药量与辅料量相差悬殊,所以在制备制剂时优选通过溶剂分散法配合分次喷洒使得恩替卡韦与辅料均匀混合的方法,有效地改善了普通混合制剂工艺所导致的含量分布不均匀的问题。(4)本发明还采用崩解剂联用技术,即内加PVPP、外加CMC-Na高效崩解剂的工艺以保证本品的分散均匀性。(5)恩替卡韦为难溶性药物,有研究表明,国产片剂与原研上市制剂相比,溶出度及生物利用度往往较低。而将恩替卡韦固体分散体进一步制成分散片后,可使本难溶于水的恩替卡韦由于预先制成了固体分散体致使其崩散后能迅速地溶解并被吸收,更快地起到抑制HBV病毒的作用,大大地提高生物利用度;同时由于其均匀分散,避免了片剂在体内瞬时局部释放造成的局部浓度过高而致的不良反应;另外还可将分散片加入牛奶或水中溶散后供有吞咽困难的病人服用,增加患者顺应性。下面结合具体实施例对本发明进行进一步的说明,但不限定本发明的保护范围。实施例及对比例中所采用原料均可通过商购得到,本发明没有特殊限定。另,以下实施例中制备的恩替卡韦制剂的剂型以分散片为例,但不仅限于。实施例1本实施例用于说明本发明提供的恩替卡韦固体分散体。取恩替卡韦0.5g,PEG-40002.0g,甘露醇0.5g,混合均匀,置高压釜中,设定制备压力为25MPa、温度为50℃、时间为12h,待釜内温度达到设定值后,通入CO2直到压力达到设定值,保持温度与压力为设定时间,减压收集釜内的固体分散体,粉碎,过筛,得到本实施例的恩替卡韦固体分散体,记为S1。对比例1物理混合制备恩替卡韦-载体物理混合物:取恩替卡韦0.5g,PEG-40002.0g,甘露醇0.5g,混合均匀,得到本对比例的恩替卡韦固体分散体,记为DS1。对比例2溶剂法制备恩替卡韦固体分散体:取恩替卡韦0.5g,PEG-40002.0g,甘露醇0.5g,混合均匀,加120ml无水乙醇搅拌使溶解,溶液置旋转蒸发仪蒸去乙醇,刮取所得固体物置真空干燥箱中干燥至恒重,粉碎,过筛,得到本对比例的恩替卡韦固体分散体,记为DS2。对比例3熔融法制备恩替卡韦固体分散体:取恩替卡韦0.5g,PEG-40002.0g,甘露醇0.5g,混合均匀,80℃条件下加热熔融,缓慢加入无水乙醇20ml,搅拌使均匀分散,持续混合2h后,真空干燥箱中干燥至恒重,粉碎,过筛,得到本对比例的恩替卡韦固体分散体,记为DS3。实施例2-5本实施例2-5用于说明本发明提供的超临界流体技术制备恩替卡韦固体分散体的优选工艺条件。以PEG为亲水性载体材料,且恩替卡韦与PEG的质量比为1:5,在压力为25MPa,温度为50℃的条件下,采用与实施例1相似的步骤制备得到实施例2-5的恩替卡韦固体分散体,不同之处在于各实施例分别设定反应时间为6h、9h、12h和24h,记为S2-S5。实施例6-11本实施例6-11用于说明本发明提供的超临界流体技术制备恩替卡韦固体分散体的优选工艺条件。在压力为25MPa,温度为50℃、反应时间为12h的条件下,采用与实施例1相似的步骤制备得到实施例2-5的恩替卡韦固体分散体,不同之处在于各实施例中的亲水性载体和恩替卡韦与亲水性载体的用量比如下表1所示,得到的恩替卡韦固体分散体记为S6-S11。表1样品载体药物载体配比样品载体药物载体配比S6甘露醇1:5S9PEG1:10S7甘露醇1:10S10PEG:甘露醇(4:1)1:5S8PEG1:5S11PEG:甘露醇(4:1)1:10实施例12-16本实施例12-16用于说明本发明提供的超临界流体技术制备恩替卡韦固体分散体的优选工艺条件。以PEG:甘露醇(质量比为4:1)为亲水性载体材料,在压力为25MPa,温度为50℃、反应时间为12h的条件下,采用与实施例1相似的步骤制备得到实施例12-16的恩替卡韦固体分散体,不同之处在于各实施例分别设定恩替卡韦与载体的比例为1:2、1:4、1:6和1:8,记为S12-S16。实施例17-20本实施例17-20用于说明本发明提供的超临界流体技术制备恩替卡韦固体分散体的优选工艺条件。以PEG:甘露醇(质量比为4:1)为亲水性载体材料,按1:6进行药物载体配比,在压力为25MPa,反应时间为12h的条件下,采用与实施例1相似的步骤制备得到实施例17-20的恩替卡韦固体分散体,不同之处在于各实施例分别设定反应温度为30℃、40℃、50℃、60℃,记为S17-S20。实施例21-23本实施例2-5用于说明本发明提供的超临界流体技术制备恩替卡韦固体分散体的优选工艺条件。以PEG:甘露醇(质量比为4:1)为亲水性载体材料,按1:6进行药物载体配比,在温度为50℃、反应时间为12h的条件下,采用与实施例1相似的步骤制备得到实施例17-20的恩替卡韦固体分散体,不同之处在于各实施例分别设定反应压力为20MPa、25MPa和30MPa,记为S21-S23。实施例24本实施例用于说明本发明提供的恩替卡韦制剂,具体为恩替卡韦分散片。本实施例制备的恩替卡韦制剂S10的处方(1000片)如下:该恩替卡韦制剂S10的制备方法如下:(1)将恩替卡韦与PEG4000、甘露醇混匀,置高压反应釜中,在压力为25MPa,温度为50℃的条件下,反应12h。取出,粉碎,得到恩替卡韦固体分散体备用;(2)混合辅料:将稀释剂、内加崩解剂均匀混合;(3)制湿颗粒:取上述恩替卡韦固体分散体,加1~5倍量的水溶解,均匀地喷洒在混合辅料上,制备软材,24目筛制粒;(4)干燥整粒:湿颗粒于55℃~60℃烘干,30目整粒;(5)总混:加入外加崩解剂及润滑剂,混合均匀;(6)压片,得到本实施例的恩替卡韦制剂,记为S10。实施例25本实施例用于说明本发明提供的恩替卡韦制剂,具体为恩替卡韦分散片。本实施例制备的恩替卡韦制剂S20的处方(1000片)如下:本实施例提供的恩替卡韦制剂S25的制备方法与实施例24的制备方法相同,在此不再赘述。实施例26本实施例用于说明本发明提供的恩替卡韦制剂,具体为恩替卡韦分散片。本实施例制备的恩替卡韦制剂S30的处方(1000片)如下:本实施例提供的恩替卡韦制剂S26的制备方法与实施例24的制备方法相同,在此不再赘述。实施例27本实施例用于说明本发明优选的溶剂分散法制备恩替卡韦制剂S27。本实施例中,制备所述恩替卡韦制剂S27所采用的原料与实施例24相同,制备方法不同,具体包括:取实施例24的制备方法步骤(1)制备所得的恩替卡韦固体分散体4.5g(相当于恩替卡韦0.5g),加水溶解,分次均匀地喷洒于混合辅料中,制备软材,过筛制粒,干燥,整粒,加外加崩解剂与润滑剂混合,压片,得到本实施例的恩替卡韦制剂S27。实施例28本实施例用于说明采用常规的普通混合法制备恩替卡韦制剂S28。本实施例中,制备所述恩替卡韦制剂S28所采用的原料与实施例24相同,制备方法不同,具体包括:取实施例24中所得恩替卡韦固体分散体4.5g(相当于恩替卡韦0.5g),加稀释剂、内加崩解剂按递加稀释法均匀混合,加水制备软材,过筛制粒,干燥,整粒,加外加崩解剂与润滑剂混合均匀,压片,得到本实施例的恩替卡韦制剂S28。性能测试(1)差示扫描量热(DSC)试验将载体(A-甘露醇;B-聚乙二醇:甘露醇(质量比为4:1)物理混合;C-聚乙二醇)、恩替卡韦原料药D、实施例1和对比例1-3制备的恩替卡韦固体分散体S1、DS1-DS3分别进行测试,测试条件为以空铝坩锅为参考池,另一空铝坩锅为样品池,放入约10mg的样品,扫描速度为10℃/min,扫描范围为0℃~300℃。各样品的DSC曲线如图1所示。由图1所示的结果可知,物理混合物DS1中,晶体仍大量存在;溶剂法、熔融法制备的恩替卡韦固体分散体DS2、DS3中,少量的仍以晶体态存在;本发明的超临界法制备的恩替卡韦固体分散体S1中,晶体几乎完全消失,可能以无定形状态或分子状态存在于固体分散体中。(2)溶出度试验(2.1)制备方法不同测试样品:恩替卡韦原料药D,恩替卡韦固体分散体S1、DS1-DS2。以900mL的PH6.8磷酸盐缓冲液为溶出介质,温度(37±0.5)℃,转速100rpm,分别于5、10、15、20、30、45、60min取样,每次2ml,同时补充同温同体积的新鲜溶出介质;样品经0.45μm微孔滤膜滤过,HPLC测定。其体外溶出曲线如图2所示。由图2的结果可知,本发明提供的超临界流体制备的恩替卡韦固体分散体D的体外溶出度得到了明显的提升。(2.2)不同反应时间测试样品:恩替卡韦固体分散体S2-S5。将各恩替卡韦固体分散体S2-S5(相当于恩替卡韦0.5mg),置于溶出度仪中,测定药物的溶出度,并绘制溶出曲线见图3。由图3的结果可知,恩替卡韦固体分散体S2-S5中药物的溶出度随着反应时间的延长有所提升。而当反应时间为12h和24h时,固体分散体中药物在60min的累积溶出度相差不多,而反应6h和9h所得固体分散体在60min的累积溶出度明显低很多。本发明从节约能耗方面考虑,最终确定超临界制备固体分散体的最佳反应时间为12h。(2.3)不同载体测试样品:恩替卡韦固体分散体S6-S11。将各恩替卡韦固体分散体S6-S11(相当于恩替卡韦0.5mg),置溶出度仪中,测定药物的溶出度,以累积溶出度为判定指标,绘制溶出度曲线,如图4所示。由图4的结果可知,以不同物质为载体材料制备出的固体分散体的溶出度具有显著差异,其中以PEG:甘露醇(4:1)为载体材料制备的固体分散体对药物的增溶效果最佳。因此本发明确定以PEG:甘露醇(4:1)为最佳的亲水性载体材料。(2.4)不同药物载体配比测试样品:恩替卡韦固体分散体S12-S16。将各恩替卡韦固体分散体S12-S16(相当于恩替卡韦0.5mg),置溶出度仪中,测定药物的溶出度,绘制溶出曲线如图5。由图5的结果可知,随着混合载体材料用量的增加,药物的溶出度也随着增大,而当药物载体的用量大于1:6时,药物的溶出度反而降低,原因可能是载体遇水产生粘性,反而阻碍了药物的溶出。因此本发明中,恩替卡韦:(PEG:甘露醇(4:1))的最佳配比为1:6。(2.5)不同反应温度测试样品:恩替卡韦固体分散体S17-S20。将各恩替卡韦固体分散体S17-S20(相当于恩替卡韦0.5mg),置溶出仪中,测定药物的溶出度,绘制溶出曲线见图6。由图6的结果可知,在一定温度范围内,恩替卡韦固体分散体的溶出度随着制备温度的升高而有所提高。其中当制备温度为50℃是,药物在60min时的累积溶出度最大。(2.6)不同反应压力测试样品:恩替卡韦固体分散体S21-S23。将各恩替卡韦固体分散体S21-S23(相当于恩替卡韦0.5mg),置溶出仪中,测定药物的溶出度,绘制溶出曲线如图7。由图7的结果可知,当制备压力为25MPa时,药物具有最大的溶解度。综上,本发明中,采用超临界流体技术制备恩替卡韦固体分散体的最佳实施例工艺为:以PEG-甘露醇(4:1)为载体材料,按1:6的比例进行药物载体配比,在制备压力为25MPa、温度为50℃的条件下,反应时间为12h。(3)不同方法制备的恩替卡韦制剂性能测试(3.1)高效液相色谱法测定含量均匀度的色谱条件与系统适用性:以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(5:95)为流动相A,乙腈为流动相B,按下表进行线性梯度洗脱。检测波长为254nm;流速为每分钟1.0ml。理论板数按恩替卡韦峰计算不低于30000,恩替卡韦与相邻的杂质峰之间的分离度应符合规定。(3.2)含量均匀度的测定方法:分别取实施例27、28制备所得恩替卡韦制剂S27、S28各10片,分别置于25ml的容量瓶中,加甲醇适量超声30min使溶解并定容,摇匀,过滤(过0.25um的微孔滤膜),既得供试品溶液,精密吸取供试品20ul,照上述色谱条件高效液相下测定。照2015版中国药典第4部0951法计算恩替卡韦制剂的含量均匀度。(3.3)溶出度的测定方法:分别取实施例27、28制备所得恩替卡韦制剂S27、S28各6片,以900mL的PH6.8磷酸盐缓冲液为溶出介质,温度(37±0.5)℃,转速100rpm,于60min时进行取样,样品经0.45μm微孔滤膜滤过,精密吸取供试品20ul,照上述色谱条件于高效液相下测定溶出度,并计算溶出均一性。结果如表2所示。表2恩替卡韦分散片两种制备工艺结果比较由上表2的结果可知,采用本发明优选的溶剂分散法和常规的普通混合法制备得到的恩替卡韦分散片的含量均匀度均符合中国药典2015年版的标准,经F检验,两者具有显著性差异(P<0.01),但采用本发明优选的溶剂分散法制得的恩替卡韦分散片分散的更均匀,且溶出的均一性更好。以上所述的本发明实施方式,并不构成对本发明保护范围的限定。任何在本发明的精神和原则之内所作的修改、等同替换和改进等,均应包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1