过饱和固体自乳化制剂及其制备方法与流程
本发明属于药物制剂技术领域,更具体地,本发明涉及一种过饱和固体自乳化制剂及其制备方法。
背景技术:
口服给药是一种最常见、最简单、患者最易接受的给药途径。药物经口服进入胃肠道之后首先需要通过溶解过程,才能被机体吸收,进而发挥药效。生物药剂学分类系统(Biopharmaceutical classification system,BCS)中的BCS II类药物具有低溶解度、高渗透性,其在消化道的溶出过程往往成为该类药物吸收的限速环节。统计数据表明,BCS II类药物在已上市药物中所占比例为40%,而在研发中药物这一比例高达70%。BCS II类药物的溶解度差导致的生物利用度低是药理活性物质不能成为治疗药物的主要原因。因此如何有效提高BCS II类药物的溶解度和溶出速率,进而改善其口服生物利用度是药学研究的重要内容。
难溶性药物固体自乳化制剂(Solid self-emulsifying drug delivery system,SSEDDS),该体系兼具自乳化技术(提高溶出和生物利用度)和固体制剂(稳定性好、储存方便、顺应性好)的双重优势,并克服了液体自乳化系统存在的诸多缺点。
然而在难溶性药物SSEDDS中,药物以无定型态存在于固相载体,由于没有晶格能的束缚,药物分子迅速溶出,自乳化成分遇水后从固相载体解吸附自发乳化形成O/W乳剂,药物分子进一步增溶于乳滴之中,导致药物在胃肠道中的表观溶解度远远超过其平衡溶解度,形成过饱和溶液。该溶液属于热力学不稳定体系,药物会自发析出晶体,使药物的表观溶解度趋向于极低的平衡溶解度,导致药物浓度降低,吸收减少,最终影响药物的体内生物利用度和药物疗效。因此维持药物在胃肠道的过饱和状态,保证药物有足够的时间经胃肠道吸收以达到提高生物利用度的目的,具有非常重要的意义。
技术实现要素:
基于此,为了克服上述现有技术的缺陷,本发明提供了一种过饱和固体自乳化制剂及其制备方法。
为了实现上述发明目的,本发明采取了以下技术方案:
一种过饱和固体自乳化制剂,由析晶抑制剂与难溶性药物固体自乳化系统(Solid self-emulsifying drug delivery system,SSEDDS)制备而成;所述难溶性药物固体自乳化系统由介孔二氧化硅SBA-15与难溶性药物液体自乳化传递系统(Self-emulsifying drug delivery system,SEDDS)制备而成,所述液体自乳化传递系统由质量比为1~2:1~4:1~4:3~6的难溶性药物、油相、乳化剂、和助乳化剂组成,所述难溶性药物液体自乳化传递系统与介孔二氧化硅SBA-15的质量比为1~4:1,所述析晶抑制剂为两亲性高分子聚合物Soluplus,所述析晶抑制剂与难溶性药物的重量比为0.2~2:1。
在其中一些实施例中,所述析晶抑制剂与难溶性药物的重量比为0.5~2:1。
在其中一些实施例中,所述析晶抑制剂与难溶性药物的重量比为1:1。
在其中一些实施例中,所述难溶性药物液体自乳化传递系统与介孔二氧化硅SBA-15的质量比为2~3:1。
在其中一些实施例中,所述液体自乳化传递系统与介孔二氧化硅SBA-15的质量比为3:1。
在其中一些实施例中,所述难溶性药物、油相、乳化剂、和助乳化剂的质量比为1.5~2:2~3:2~3:3~4。
在其中一些实施例中,所述难溶性药物、油相、乳化剂和助乳化剂的质量比为1.5:2.55:2.55:3.4。
在其中一些实施例中,所述难溶性药物为BCS II类药物,其溶解度低,脂溶性高。
在其中一些实施例中,所述难溶性药物为非诺贝特、塞来昔布、或卡马西平。
在其中一些实施例中,所述油相为单辛酸甘油酯、油酸、油酸乙酯、中链甘油三酸酯、三甘油辛酸/癸酸酯、丙烯基乙二醇月癸酸酯中的一种或几种。
在其中一些实施例中,所述乳化剂为Tween 80、聚氧乙烯油酸酯、乙氧基聚氧乙烯甘油酯、聚乙二醇甘油酯中的一种或几种。
在其中一些实施例中,所述助乳化剂为乙醇、丙二醇、聚乙二醇(PEG)、异丙醇、甘油、乙二醇单乙基醚(Transcutol)、二甲基异山梨酯中的一种或几种。
在其中一些实施例中,所述液体自乳化传递系统由难溶性药物、油酸乙酯、Cremophor RH40和Transcutol HP组成。
本发明还提供了上述过饱和固体自乳化制剂的制备方法,采取了以下具体技术方案:
一种过饱和固体自乳化制剂的制备方法,包括以下步骤:
(1)将油相、乳化剂、助乳化剂混合均匀,得到均一澄清的油状溶液;将难溶性药物加入上述油状溶液中,混合均匀后,于30~40℃水浴平衡待药物完全溶解,即得该难溶性药物的液体自乳化传递系统,所述难溶性药物、油相、乳化剂和助乳化剂的质量比为1~2:1~4:1~4:3~6;
(2)将步骤(1)的难溶性药物的液体自乳化传递系统,加入无水乙醇,室温搅拌下,加入介孔二氧化硅SBA-15,保持搅拌20~30h使其达到吸附平衡,于35~50℃烘干除去溶剂,即得难溶性药物固体自乳化系统,所述难溶性药物的液体自乳化传递系统与介孔二氧化硅SBA-15的质量比为1~4:1;
(3)按析晶抑制剂与难溶性药物重量比为0.2~2:1的比例,将析晶抑制剂加入上述难溶性药物固体自乳化系统中,即得所述过饱和固体自乳化制剂。
与现有技术相比,本发明具有以下有益效果:
1、本发明的过饱和固体自乳化制剂通过加入析晶抑制剂Soluplus,从热力学角度讲,Soluplus通过增加难溶性药物的平衡溶解度,降低了体系过饱和度,减小了药物的结晶驱动力;从动力学角度讲,Soluplus与难溶性药物之间可能存在较强的氢键作用,发挥抑制和延缓结晶的效应;析晶抑制剂Soluplus通过抑制过饱和难溶性药物的成核与晶体生长速率来达到抑晶效果,维持药物溶液的过饱和状态,保证难溶性药物的吸收,生物利用度高,有望开发成产品,解决临床上难溶性药物生物利用度低导致的低药效问题,具有临床应用的实际价值;
2、本发明的过饱和固体自乳化制剂的制备方法工艺简单,具有良好的开发和应用前景。
附图说明
图1为本发明试验例1中非诺贝特过饱和浓度-时间曲线(A)与过饱和浓度-时间曲线下面积AUCss(B);
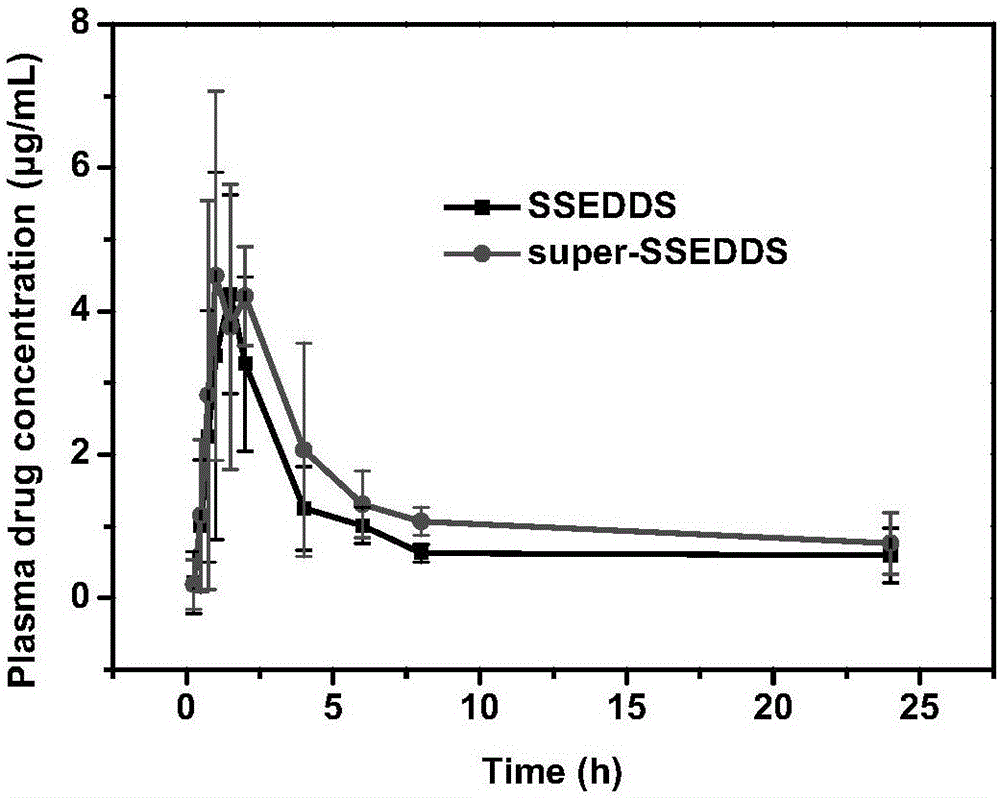
图2为本发明试验例3中不同的非诺贝特制剂在Beagle犬体内的血药浓度-时间变化曲线,其中,super-SSEDDS为FNB过饱和固体自乳化制剂,SSEDDS为未加析晶抑制剂的FNB固体自乳化制剂。
具体实施方式
下面结合附图和具体实施例进一步叙述本发明,本发明未述及之处适用于现有技术。下面给出本发明的具体实施例,但实施例仅是为了进一步详细叙述本说明,并不限制本发明的权利要求。
以下实施例中,如无特殊说明,原料均来源于市售。
实施例1非诺贝特过饱和固体自乳化制剂
本实施例的非诺贝特过饱和固体自乳化制剂的构建方法包括以下步骤:
以非诺贝特(FNB)为难溶性药物,油相为油酸乙酯,乳化剂为Cremophor RH40,助乳化剂为Transcutol HP。其制备方法包括以下步骤:
分别称取油相2.55g、乳化剂2.55g、助乳化剂3.4g混合,得到均一澄清的油状溶液,称取1.5g难溶性药物FNB加入上述溶液溶解,即得该难溶性药物液体SEDDS。称取3.0g上述难溶性药物液体SEDDS,加入10mL无水乙醇,加入1.0g固化载体SBA-15,烘干除去溶剂,即得该难溶性药物SSEDDS。称取析晶抑制剂两亲性高分子聚合物Soluplus 1.0g加入等同于1.0g FNB的上述难溶性药物SSEDDS中,即得本实施例的非诺贝特过饱和固体自乳化制剂。
实施例2塞来昔布过饱和固体自乳化制剂
本实施例的塞来昔布过饱和固体自乳化制剂的构建方法包括以下步骤:
以塞来昔布为难溶性药物,油相为油酸乙酯,乳化剂为Cremophor RH40,助乳化剂为Transcutol HP。其制备方法包括以下步骤:
分别称取油相3.0g、乳化剂3.0g、助乳化剂4.0g混合,得到均一澄清的油状溶液,称取2.0g难溶性药物塞来昔布加入上述溶液溶解,即得该难溶性药物液体SEDDS。称取3.0g上述难溶性药物液体SEDDS,加入10mL无水乙醇,加入1.0g固化载体SBA-15,烘干除去溶剂,即得该难溶性药物SSEDDS。称取析晶抑制剂两亲性高分子聚合物Soluplus0.8g加入等同于1.0g塞来昔布的上述难溶性药物SSEDDS中,即得本实施例的塞来昔布过饱和固体自乳化制剂。
实施例3卡马西平过饱和固体自乳化制剂
本实施例的卡马西平过饱和固体自乳化制剂的构建方法包括以下步骤:
以卡马西平为难溶性药物,油相为油酸乙酯,乳化剂为Cremophor RH40,助乳化剂为Transcutol HP。其制备方法包括以下步骤:
分别称取油相2.5g、乳化剂2.5g、助乳化剂3.5g混合,得到均一澄清的油状溶液,称取1.5g难溶性药物卡马西平加入上述溶液溶解,即得该难溶性药物液体SEDDS。称取3.5g上述难溶性药物液体SEDDS,加入10mL无水乙醇,加入1.0g固化载体SBA-15,烘干除去溶剂,即得该难溶性药物SSEDDS。称取析晶抑制剂两亲性高分子聚合物Soluplus1.5g加入等同于1.0g卡马西平的上述难溶性药物SSEDDS中,即得本实施例的卡马西平过饱和固体自乳化制剂。
实施例4非诺贝特过饱和固体自乳化制剂
本实施例的非诺贝特过饱和固体自乳化制剂的构建方法包括以下步骤:
以非诺贝特(FNB)为难溶性药物,油相为油酸乙酯,乳化剂为Cremophor RH40,助乳化剂为Transcutol HP。其制备方法包括以下步骤:
分别称取油相2.6g、乳化剂2.4g、助乳化剂3.3g混合,得到均一澄清的油状溶液,称取1.7g难溶性药物FNB加入上述溶液溶解,即得该难溶性药物液体SEDDS。称取2.9g上述难溶性药物液体SEDDS,加入10mL无水乙醇,加入1.0g固化载体SBA-15,烘干除去溶剂,即得该难溶性药物SSEDDS。称取析晶抑制剂两亲性高分子聚合物Soluplus0.6g加入等同于1.0gFNB的上述难溶性药物SSEDDS中,即得本实施例的非诺贝特过饱和固体自乳化制剂。
试验例1实施例1的非诺贝特过饱和固体自乳化制剂的体外过饱和试验
精密称取等同于FNB100mg的实施例1的非诺贝特过饱和固体自乳化制剂,过饱和溶出介质为200mL去离子水,转速100rpm,温度37±0.5℃,分别于0,1,2,3,4,5,6h取样2mL。用0.22μm聚四氟乙烯滤膜过滤,取续滤液0.5mL,立即采用无水乙醇稀释至10mL,在286nm处测定紫外吸收,计算各个时间点非诺贝特浓度。平行三份,以消除误差。
图1A和B分别为过饱和浓度-时间变化曲线与过饱和浓度-时间曲线下面积AUCss,结果表明,当Soluplus与FNB的质量比由0增加至1时,AUCss随着Soluplus用量的增加而逐渐增加,表明在此范围内,FNB的结晶速率逐渐减慢,Soluplus维持过饱和状态的能力随着其用量的增加而增大。当Soluplus与FNB的质量比由1增加至10时,AUCss随着Soluplus用量的增加而无显著性变化,表明在此范围内,FNB的结晶速率基本不变,Soluplus维持过饱和状态的能力保持恒定。故在过饱和固体自乳化体系中,确定析晶抑制剂Soluplus与FNB的质量比为1:1。
试验例2实施例1的非诺贝特过饱和固体自乳化制剂重新乳化试验
按照实施例1的方法构建非诺贝特液体自乳化体系。取等同于0.3mL液体自乳化的过饱和固体自乳化制剂,加入200mL水,转速为50rpm,待其完全乳化,观察乳剂外观。所得样品静置过夜,取上清液进行粒径测定。表1为固体自乳化及过饱和固体自乳化体系的粒径及多分散系数。
表1固体自乳化及过饱和固体自乳化体系的粒径及多分散系数(n=3)
表1结果表明,Soluplus加入后,乳滴的粒径略有增加,可能是由于部分Soluplus的高分子链段嵌入乳滴的表面,造成了一定的水化作用,从而增加了乳滴的空间位阻效应。两种粒径的PDI均小于0.20,表明乳剂的粒度均一,析晶抑制剂Soluplus的加入并未对体系的乳化行为产生显著性影响。
试验例3实施例1的非诺贝特过饱和固体自乳化制剂的Beagle犬体内药动学研究
受试制剂为实施例1中制备的析晶抑制剂Soluplus与FNB的质量比为1:1的FNB过饱和固体自乳化制剂super-SSEDDS和未加析晶抑制剂的FNB固体自乳化制剂SSEDDS。
采取两制剂双交叉实验方案设计,6只Beagle犬随机分为两组,每组三只,用于体内药动学研究。实验前禁食(不禁水)12h,清洗期为两周。每只Beagle犬分别给予相当于100mgFNB的固体自乳化制剂、过饱和固体自乳化制剂。给药后在固定时间点(0、0.25、0.50、0.75、1、1.5、2、3、4、6、8、24h)于后肢股静脉取血4mL于含肝素钠的负压取血管中,混合均匀后5000rpm,离心5min,取上层血浆,于-20℃冰箱保存以待分析。取含药血浆样品0.5mL,精密加入50μL的内标吲哚美辛工作液(50μg/mL),200μL浓度为1mol/L的盐酸溶液,于涡旋器上振荡混匀5min。再加入4mL无水乙醚,涡旋5min,8000rpm离心5min,取上清液,经氮吹仪浓缩挥干溶剂。加入200μL流动相复溶,涡旋5min,经16000rpm离心5min,取上清液,采用高效液相色谱分析FNB酸的血液浓度。
色谱条件:色谱柱:Phenomenex C18(250mm×4.6mm,5μm);流动相:乙腈:0.1%醋酸=65:35;检测波长:286nm;流速:1mL/min;柱温:40℃;进样量:50μL。
图2为不同的FNB制剂在Beagle犬体内的血药浓度-时间变化曲线结果表明,两种制剂的血药浓度-时间曲线变化趋势一致,过饱和制剂的血药浓度高于固体自乳化制剂。
进一步采用WinNonlin V3.3软件计算各制剂的药动学参数,将Beagle犬体内血药浓度-时间曲线结果进行房室模型拟合,结果显示房室模型的拟合系数较小,且各组内Beagle犬的房室模型不一致,因此本发明最终采用非房室模型进行拟合,计算药动学参数。表2为Beagle犬口服给药后血浆药物浓度的药动学参数。
表2 Beagle犬口服给药后血浆药物浓度的药动学参数(n=5)
表2结果表明,SSEDDS及super-SSEDDS的Cmax、Tmax无显著性差异,super-SSEDDS的AUC0→∞高于SSEDDS,表明在固体自乳化体系中加入Soluplus,可进一步提高FNB的吸收。在固体自乳化体系中,模型药物FNB以无定型态分布于自乳化骨架中,由于没有晶格束缚,药物的溶出速率较快。同时,本发明采用的固化载体材料SBA-15具有较高的比表面积,有利于水分子的进入,进一步增加了药物及自乳化成分的释放。药物的迅速释放,导致胃肠道中药物的表观溶解度远远超过其平衡溶解度,析晶抑制剂Soluplus的加入,维持了体系的过饱和状态,保证了FNB的吸收。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!