一种引物组合物及其应用的制作方法
本发明涉及生物
技术领域:
,尤其涉及一种引物组合物及其应用。
背景技术:
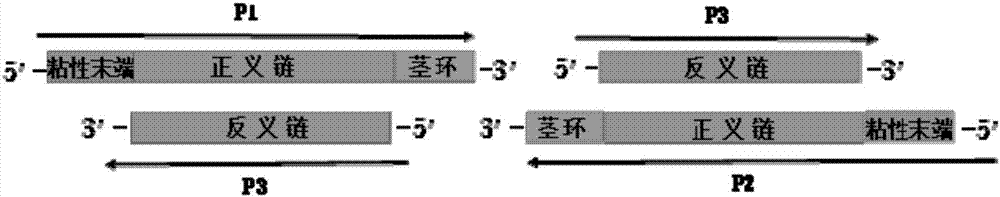
:dna甲基化转移酶1(dnamethyltransferase1,dnmt1)是细胞内dna进行复制修复,并维持正常甲基化的关键酶。dnmt1的主要功能是保持细胞内某些蛋白质或核酸甲基化状态,具体以活性甲基化合物(如s-腺苷基甲硫氨酸)为甲基供体,将甲基转移到特定的分子上。甲基化修饰与细胞内多种过程相关,包括基因表达的调控、蛋白质功能的调节、核糖核酸加工以及重金属修饰等。dnmt1在结构上包含三个区域,即c端的催化区、n端的靶区域以及功能未知区域。研究表明,dnmt1的异常表达可能是导致体内甲基化状态改变的重要原因,并与衰老、老年痴呆及多种肿瘤等疾病的形成密切相关。当前,靶向抑制dnmt1基因的表达已经成为新药研究的热点。研究调节基因表达方法,特别是抑制基因表达的一种重要技术方法是构建shrna载体,并使其在细胞中表达,从而通过rna干扰机制沉默基因的表达。shrna(shorthairpinrna)即“短发夹rna”,包括两个短反向重复序列,中间由茎环(loop)序列分隔的,组成发夹结构,随后再连上5-6个t作为rna聚合酶ⅲ的转录终止子。shrna载体构建是通过设计目的基因shrna序列,并克隆至特定载体中;经过转染细胞后转录生成shrna,利用细胞内dicer酶生成小片段干扰rna,并最终靶向目的基因mrna,从而达到基因沉默的效果。cn103757021a公开了一种沉默dnmt基因的双链sirna分子及其应用,针对人肿瘤细胞dnmt基因的核苷酸序列设计合成sirna,所述sirna转入肝癌细胞后能明显促进免疫原性相关蛋白的表达。该发明的sirna分子可以应用于制备调节细胞中dnmt基因的药物中发挥rna干扰的作用,增强免疫细胞的毒性反应,诱导肿瘤细胞消退,达到治疗肿瘤的目的。cn105132413a公开一种用于制备shrna的引物及制备方法、载体及构建方法,用于制备shrna的引物包括引物p1、p2、p3;p1长度为34nt,从5’端开始,依次为突出的粘性末端accg,21nt的sirna正向序列,loop序列,以及与sirna正向序列3’末端3个碱基反向互补的碱基;p2的5’端4个碱基由accg改变为aaaa,其余序列与p1相同;p3是sirna正向序列5’末端18nt序列的反向互补序列。目前构建shrna载体的首要步骤是根据目的基因设计两条寡聚核苷酸(oligo),每条寡聚核苷酸都包含:正向引物-茎环-反义链的结构,正向引物和反义链是两个反向重复序列,长度通常为21bp,而茎环结构通常长度为6bp,再加上两点设计的粘性末端,因此每一条寡聚核苷酸长度通常在55bp以上。基于当前的设计方法,从合成角度看,长的核苷酸链会增加合成难度,更容易产生突变,不利于研究工作的开展甚至导致基因沉默的效果不佳;而从经济的角度看,越长的核苷酸链合成价格更高,增加了研究工作的经济成本。因此,研究一种更为高效的构建shdnmt1载体的方法,即引物组合物的设计方法,降低合成的难度和成本,减少合成引物的突变,有效抑制dnmt1的表达,将shdnmt1作为基因治疗手段,研究shdnmt1在肺癌等肿瘤疾病的基因治疗方面的应用,具有十分重要的意义。技术实现要素:针对现有技术的不足及实际的需求,本发明提供一种引物组合物及其应用,本发明将shrna载体构建过程中根据目的基因设计的两条寡聚核苷酸引物拆分成三条,缩短了引物长度,降低了合成难度和成本,不容易产生突变,提高了shrna载体的准确性。由所述引物组合物制备得到的shrna、包含该shrna的慢病毒载体和药物能够抑制目的基因的表达,用于癌症的治疗和药物的研发。为达此目的,本发明采用以下技术方案:一方面,本发明提供一种引物组合物,所述引物组合物包括p1、p2和p3,所述p1包括粘性末端1、正向引物和茎环序列,所述p2包括粘性末端2、反向引物和所述p1的茎环序列的反向互补序列,p3为正向引物的反向互补序列,其中,所述正向引物为目的基因的sirna对应的靶位点序列,所述正向引物和所述反向引物互为互补链。发明人在长期的shrna实验过程中,出于缩短引物长度以减少突变和降低成本的动机,反复摸索实验条件和步骤,创造性地将shrna载体构建过程中惯用的引物设计原则进行更改,把两条引物拆分成三条,经实践后证明真实有效,能够显著抑制基因的表达。优选地,所述正向引物的目的基因包括,优选为dnmt1。优选地,所述dnmt1的sirna对应的靶位点核苷酸序列如seqidno:1所示,所述seqidno:1为ggagaacggtgctcatgctta。优选地,所述p1的核苷酸序列如seqidno:2所示,所述seqidno:2为agcgggagaacggtgctcatgcttatagtgaagccacagatgta。优选地,所述p2的核苷酸序列如seqidno:3所示:所述seqidno:3为ggcaggagaacggtgctcatgcttatacatctgtggcttcacta。优选地,所述p3的核苷酸序列如seqidno:4所示,所述seqidno:4为taagcatgagcaccgttctcc。第二方面,本发明提供一种shrna,所述shrna由第一方面所述的引物组合物制备得到,所述shrna的序列如seqidno:1所示,所述seqidno:1为ggagaacggtgctcatgctta。第三方面,本发明提供一种如第二方面所述shrna的制备方法,包括如下步骤:(1)将p1、p2和p3用te缓冲液溶解;(2)加超纯水稀释至终浓度为0.1-1μm,例如可以是0.1μm、0.3μm、0.5μm、0.8μm、0.9μm或1μm,优选为0.1μm;(3)置于沸水浴中反应1min,自然冷却至室温,获得shrna。第四方面,本发明提供一种包含第二方面所述shrna的慢病毒载体。第五方面,本发明提供一种如第四方面所述慢病毒载体的构建方法,包括如下步骤:将合成的shrna序列连接到慢病毒载体上,转化至感受态细胞后培养过夜。优选地,所述慢病毒载体为plvx-u6-ccdb-ef1-copgfp-pgk-puro。优选地,所述感受态细胞为stbl3。第六方面,本发明提供一种药物组合物,包含如第二方面所述的shrna和/或第四方面所述的慢病毒载体。第七方面,本发明提供一种如第六方面所述的药物组合物用于制备抑制dnmt1基因的表达的生物抑制剂。第八方面,本发明提供一种如第六方面所述的药物组合物用于制备治疗癌症的药物。所述癌症包括肺癌、肝癌、胃癌、乳腺癌或卵巢癌中的任一种或至少两种的组合。与现有技术相比,本发明具有如下有益效果:(1)本发明提供的引物组合物将shrna载体构建过程中惯用的引物设计原则进行修改调整,把两条引物拆分成三条,降低合成引物过程的突变率,提高shrna的准确性,降低合成难度和经济成本;(2)经退火连接制备成shrna、包含该shrna的慢病毒载体和药物,能有效抑制基因的表达,抑制癌症的增殖。附图说明图1是现有技术基于两条寡核苷酸链构建shrna载体的引物设计示意图;图2是本发明基于三条寡聚核苷酸链构建shrna载体的引物设计示意图;图3是本发明实施例3中a549细胞的dnmt1蛋白表达图,其中,β-tubulin为内参蛋白,plvx-shcontrol为针对非靶基因序列的对照shcontrol慢病毒载体及慢病毒,plvx-shdnmt1为基因dnmt1慢病毒载体及慢病毒;图4是本发明实施例4中a549细胞的细胞增殖图,其中,hoechet为染核的染料,edu(5-乙炔基-2’脱氧尿嘧啶核苷)是一种胸腺嘧啶核苷类似物。具体实施方式为更进一步阐述本发明所采取的技术手段及其效果,以下结合附图并通过具体实施方式来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。实施例1针对dnmt1基因设计引物合成shrna针对人dnmt1基因(geneid:1786)设计sirna对应的靶位点序列和引物序列;靶位点序列如seqidno:1所示,seqidno:1的序列为ggagaacggtgctcatgctta;引物序列包括:引物序列shdnmt1-p1如seqidno:2所示,seqidno:2的序列为agcgggagaacggtgctcatgcttatagtgaagccacagatgta;引物序列shdnmt1-p2如seqidno:3所示,seqidno:3的序列为ggcaggagaacggtgctcatgcttatacatctgtggcttcacta;引物序列shdnmt1-p3如seqidno:4所示,seqidno:4的序列为taagcatgagcaccgttctcc。将上述三条引物用te缓冲液溶解成10μm浓度溶液,按照表1所示体系将上述引物均匀混合后,至于沸水中反应1min,然后在水浴中自然冷却至室温。表1shdnmt1引物形成双链的退火体系试剂用量(μl)shdnmt1-p12shdnmt1-p22shdnmt1-p32ddh2o194总体积200实施例2构建靶向抑制dnmt1基因表达的shrna慢病毒载体以plvx-u6-ccdb-ef1-copgfp-pgk-puro为shrna慢病毒载体,用bsmbi(neb)进行酶切,用胶回收试剂盒(e.z.n.agelextractionkit,omgega)进行片段回收,纯化作为载体片段。将上述退火形成的shdnmt1作为插入片段,将插入片段与载体片段利用t4连接酶(promega)与16℃连接2h,然后将连接产物转化至大肠杆菌感受态stbl3,37℃过夜培养后挑取生长良好的单菌落在含有载体相应抗生素的lb培养基中扩增,提取质粒后进行测序验证,获得dnmt1的shrna慢病毒载体plvx-u6-shdnmt1-ef1-copgfp-pgk-puro。实施例3shrna慢病毒载体对dnmt1基因的抑制效果使用实施例2制备的shdnmt1慢病毒载体,在a549细胞上对目的基因dnmt1进行沉默效果的验证,具体包括以下步骤:1.shrna慢病毒载体的慢病毒包装:1)将培养的hek293细胞接种于100mm培养皿,培养12-24h,使细胞密度达到80-90%;2)利用lipo2000转染试剂,混合17μg病毒包装质粒,以及6μgshdnmt1慢病毒重组质粒,然后将转染试剂dna混合液滴加至100mm培养皿中,37℃培养箱中培养8-12h,更换新鲜培养基;3)继续培养48h后收集上清,35000rpm离心10min,收集的上清液及为慢病毒。2.shrna慢病毒感染a549细胞:1)将a549细胞终于6孔板,细胞密度为1.0×105个细胞/孔;2)培养12-24h后,分别用500μldmem+500μlshrna慢病毒+0.5μlpolybrene的质粒混合液感染a549细胞。以针对非靶基因序列的对照shcontrol慢病毒载体及病毒,作为基因dnmt1慢病毒载体及慢病毒的对照,并感染a549细胞;3)感染病毒后24h更换新鲜培养液;4)细胞感染病毒72h后,观察荧光,并加入终浓度为1μg/μl的嘌呤霉素继续培养,筛选稳定感染shdnmt1慢病毒的细胞系。3.蛋白杂交检测蛋白质水平的表达:1)用ripa裂解液稳定感染shdnmt1、shcontrol慢病毒的a549细胞,收集蛋白样品并定量,取40μg蛋白样品上样,在100v恒压进行sds-page电泳;2)转膜:250ma恒流3.0h,转膜后于5%脱脂奶粉封闭液中封闭1h,再用tbst洗膜三次。3)置于用3%bsa配制的dnmt1一抗中,4℃过夜孵育。4)第二天用tbst将膜洗三次,每次10min,加入用3%bsa配制的兔二抗,室温杂交1h后进行化学发光显影,结果如图3所示。由图3可知,与对照组相比,shdnmt1慢病毒感染的a549细胞中,dnmt1蛋白表达量明显降低,说明本发明设计的shrna引物对dnmt1基因表达有抑制效果。实施例4shrna慢病毒抑制a549细胞的增殖为探讨抑制dnmt1对a549细胞的调控作用,在a549细胞上进行慢病毒感染实验,分别感染本发明设计的shcontrol慢病毒和shdnmt1慢病毒,然后通过细胞增殖标志蛋白pcna和edu细胞增殖检测确定细胞的增殖水平。使用edu细胞增殖检测试剂盒(锐博生物)进行edu标记,根据试剂盒说明书进行实验操作,主要步骤如下:1)细胞转染:接种1×104个细胞于48孔板,培养24h后进行慢病毒感染实验,第二天培养24h后更换新鲜培养基以及20μmedu继续4小时;2)细胞染色拍照:将细胞用4%多聚甲醛室温固定30分钟,0.5%tritonx-100进行膜通透10分钟,pbs清洗,然后每孔加入150μl1×apollo染色也孵育30分钟。dna用1×hochest(每孔150μl)染色5分钟,最后在荧光显微镜下观察,结果如图4所示。由图4可知,与慢病毒对照相比,感染shdnmt1慢病毒后a549增殖下降35%。综上所述,本发明提供的引物组合物制备得到的shrna慢病毒载体能够显著抑制基因表达和癌症细胞的增殖,证明了三条引物的可行性,将引物缩短后能减少合成难度和突变,降低合成成本,提供shrna的准确性。申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属
技术领域:
的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。序列表<110>中国科学院深圳先进技术研究院<120>一种引物组合物及其应用<130>201711600ec<141>2017-11-14<160>4<170>siposequencelisting1.0<210>1<211>21<212>dna<213>人工合成()<400>1ggagaacggtgctcatgctta21<210>2<211>44<212>dna<213>人工合成()<400>2agcgggagaacggtgctcatgcttatagtgaagccacagatgta44<210>3<211>44<212>dna<213>人工合成()<400>3ggcaggagaacggtgctcatgcttatacatctgtggcttcacta44<210>4<211>21<212>dna<213>人工合成()<400>4taagcatgagcaccgttctcc21当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种荧光定量PCR技术检测意大利蜜蜂王浆主蛋白MRJP 2基因表达的方法与流程
- 一种用于对不同银杏进行基因分型的SNP引物及检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种基于荧光PCR检测ESR1基因突变的引物及探针的制造方法与工艺
- 转基因甜菜GTSB77品系特异性实时荧光PCR检测引物、探针、方法和试剂盒与流程
- 一种基于单核苷酸多态性的葡萄球菌种水平鉴定和菌株分型一体化方法与流程
- 一套毛花猕猴桃和中华猕猴桃物种间杂交子代的鉴定引物的制造方法与工艺
- 检测黄杉大小蠹的引物、试剂盒及检测方法与流程
- 环介导等温扩增技术检测铜绿假单胞菌的引物和试剂盒的制造方法与工艺
- 一种用于鉴定浙贝母的引物对及其应用的制造方法与工艺