一种密闭式慢病毒载体培养装置和培养方法与流程
本发明涉及一种密闭式慢病毒载体培养装置和培养方法。
背景技术:
哺乳动物蛋白质的转基因表达可能将正常的体细胞转化为肿瘤发生的状态,例如p53和Rb路径的失活会导致显负性p53蛋白(p53DD)的表达,一个激活细胞周期蛋白依赖性激酶(CDK)/细胞周期蛋白复合体(cyclinD1/CDK4R24C)与原癌基因的激活,Ras,和hTERT通路通过致癌的表达形式的原癌基因(c-MycT58A),H-Ras(H-RasG12V)和hTERT,可以驱动不同的人类细胞转化致瘤的状态。研究报道称使用上述的致癌基因组合可以使正常细胞发生致癌细胞转化,这表明正常的肝细胞可能用于基因工程以帮助研究发现在人体癌症的正常转化过程,当它回到宿主肝细胞中时可以形成肿瘤。
慢性病毒载体可以转导不可复制细胞同时需要编码肿瘤细胞,细胞培养实验需要在生物安全等级为3级的密闭实验室中进行。然而目前的生物医学试验所使用的培养装置仍然不能很好的保护试验人员。
技术实现要素:
为了解决目前技术中存在的问题,本发明提出了一种密闭式慢病毒载体培养装置和培养方法,该发明为密闭系统,不会产生慢病毒气溶胶,由于不会直接与慢病毒培养液接触,可有效保护试验人员。
为了实现上述技术目的,达到上述技术效果,发明是通过以下技术方案实现的:
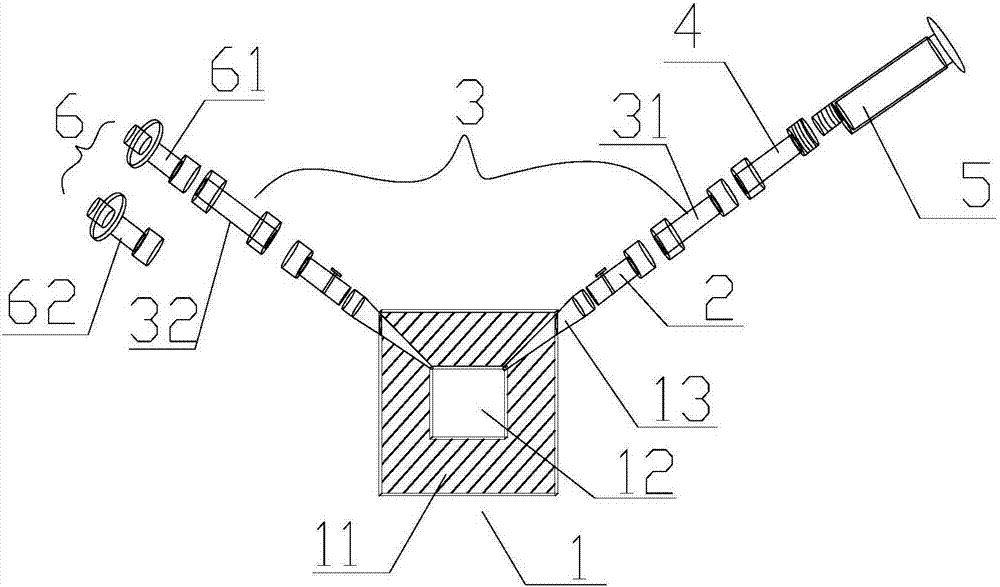
一种密闭式慢病毒载体培养装置和培养方法,培养装置由盒体、阀门、连接管、Q-syte分隔膜密闭式无针接头、Luer-Lock注射器、过滤器和密闭栓组成;
所述的盒体由上盒片和下盒片紧密贴合组成,上盒片和下盒片固定于刚性框架下,上盒片和下盒片中心部位向盒体外侧凹入形成矩形容积腔,在盒体的左右两侧各有一个变截面圆柱状通道,通道内固定有变截面圆柱状的母槽连接管;
所述的Luer-Lock注射器的接头为螺旋接头。
2、一种密闭式慢病毒载体培养装置和培养方法,慢性病毒载体培养方法包含以下步骤:
步骤一、试剂的制备:
(1)转导质粒的制备:首先使用PCR对hTERT+p53DD、cyclinD1+CDK4R24C和c-mycT58A+HRasG12V进行增强处理,在CMVV5-LUC受体质粒上的XmaI和KpnI部位之间克隆三个插入序列,并对插入序列进行荧光素酶标记基因操作,得到荧光素标记的pLenti-hTERT+53DD、pLenti-cyclinD1+CDK4R24C和pLenti-c-mycT58A+HRasG12V质粒,使用荧光素酶pLenti-CMVV5作为控制荧光素酶报告基因的控制质粒;
(2)293T细胞和Huh7.5.1细胞的细胞培养基(CM)制备:向细胞培养基中添加体积比为10%的灭活胎牛血清,体积比为1%的非必需氨基酸,10ug/mL的庆大霉素;使用0.22um的过滤器过滤后在4℃的环境中保存最多两个月;
(3)原代肝细胞(PPH)培养基(WM)制备:向500mL的William’s E培养基添加10mL的质量体积比为7.5%的牛血清蛋白,5mL的ITS-G,10-7M的地塞米松溶液,体积比为1%的非必需氨基酸,体积比为1%的青霉素和链霉素的混合溶液,体积比为1%的稳定性谷氨酸;使用0.22um的过滤器过滤后保存在-80℃的环境中;
(4)原代肝细胞(PPH)离析:将肝切成小块组织立即置于含冰块的生理盐水中,在切割表面上插入可视装置,然后在pH为7.4和37℃的环境中使用500mL的缓冲液对肝组织进行灌洗,缓冲液中包含8.3g/L的NaCl、0.5g/L的KCl、2.4g/L的HEPES,0.19g/L的EGTA;使用包含100g的IV型胶原酶、3.9g/L的NaCl、0.5g/L的KCl、2.4g/L的HEPES和0.7g/L的CaCl2·H2O的400mL缓冲液在37℃的环境中进行第二次灌洗;使用IV型的胶原酶循环灌注,然后移除大块的肝组织,轻轻摇动存储在冰水中的包含有2.4g/L的HEPESD的HBSS缓冲液即可得到靶细胞;将含有靶细胞的悬浮液通过尼龙网过滤后在4℃的环境中冲洗三次;使用Trypan试剂排斥实验验证得到的肝细胞的可用性;立即使用合格的细胞或者冻结在-80℃的添加有10%的胎牛血清溶液中以代用;
步骤二、在密闭式慢病毒载体培养装置中得到慢病毒上清液:
(1)培养装置和293T细胞的准备工作:将阀门分别连接培养装置的两侧,在阀门上连接两端为公槽的连接管,左侧的连接管上连接0.22um的过滤器,右侧的连接管连接Luer-Lock注射器;50mL的试管中含有12mL的CM培养基,将293T细胞数量倍增至3.0~7.5×106,使用Luer-Lock注射器吸入2mL空气后吸入10mL的细胞悬浮液;
(2)慢病毒上清液制备过程:向培养装置中第一次注入10mL的293T细胞悬浮液,然后将培养装置置于37℃和浓度为5%的CO2的条件下孵化6-8小时;使用注射器将培养装置里的细胞悬浮液抽出,然后再次向培养装置中注入10mL的细胞悬浮液,将培养装置置于37℃和浓度为5%的CO2的条件下孵化24小时;向培养装置后注入10mL的CM培养基后使用注射器抽出悬浮液,再次注入10mL的CM培养基,在37℃和浓度为5%的CO2的条件下孵化1小时;向1.5mL的Eppendorf试管中加入500uL的无菌水、8.1ug的HIV-gap-pol质粒、8.1ug的pbabe-hTERT+p53DD、pbabe-cyclinD1+CDK4R24C或者pbabe-cmycT58A+HRasG12V质粒、2.7ug的pMD2 VSV-G-Env质粒和62uL的CaCl2 2M;向5mL的试管中添加500uL的HBS 2x并轻微旋转,旋转在室温条件下孵化20分钟,然后添加9mL的CM培养基;将含有2mL气体和10mL的转染混合物注入培养装置中,然后注入CM培养基,在37℃和CO2浓度5%的环境中孵化24小时;使用注射器注入10mL的CM培养基然后抽出,然后再注入10mL的CM培养基,将培养装置置于在37℃和CO2浓度5%的环境中孵化24小时;将培养装置左侧0.22um的过滤器更换为0.45um的过滤器,同时过滤器连接第二个培养装置,在第二个培养装置的右侧连接0.22um的过滤器,第一个培养装置连接注射器,使用充满空气的注射器将第一个培养装置的细胞悬浮液转移到第二个培养装置中,将第二个培养装置上的所有连接管拆下,即可得到慢病毒上清液,将得到的上清液培养置于-80℃的条件下直接冻结;
步骤三、在密闭式慢病毒载体培养装置中对原代肝细胞(PPH)转染:
将PPH悬浮液注入培养装置中,置于37℃和浓度5%的CO2环境中孵化4小时,然后将培养装置正反面调换,调换后置于37℃和浓度5%的CO2环境中孵化4小时;将步骤二得到的慢病毒载体置于37℃的水中解冻,将含有肝细胞悬浮液的培养装置与慢病毒上清液的培养装置连接一起;使用注射器抽入空气后连接到慢病毒培养装置上,将慢病毒上清液转移到PPH细胞悬浮液中,将装置置于37℃和CO2浓度5%的环境中48小时;将孵化得到的转染慢病毒的PPH细胞悬浮液中注入添加有10%胎牛血清的低温贮藏介质,将得到的转导PPH细胞悬浮液培养装置置于-80℃低温下冻结。
优选的,所述的上下盒片面积为25cm2,上下盒片为透气和透明的材料制成。
本发明的有益效果是:提出了一种密闭式慢病毒载体培养装置和培养方法,该发明所用的装置可隔绝试验内部与外部的环境,不会产生慢病毒气溶胶,在密闭的培养环境中操作可避免与慢病毒培养液直接接触,可有效防止试验人员感染;同时提出的培养方法得到的慢病毒载体与传统培养方法得到的载体有相同的转导效率和转染效果。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是密闭式慢病毒载体培养装置结构图;
图2是所述的培养装置的部件连接示意图;
图3是所述的两个培养装置的连接示意图;
图4是3天后使用OCS系统和CCS系统进行培养后致癌基因编码LVs转导效率对比图,图中纵坐标为生物荧光酶活性,横坐标为不同的编码的致癌基因;
图5是使用OCS系统和CCS系统转导PPH效果对比图,图中纵坐标为生物荧光酶活性;
图6是缺失表达能力的质粒和正常的质粒感染PPH效果对比图;
图7是注入慢病毒载体的小鼠在第0天、第2天和第6天的荧光素酶活性检测图。
附图中,各标号所代表的部件列表如下:
1-盒体,11-盒片,12-容积腔,13-母槽连接管,2-阀门,3-连接管,4-Q-syte接头,5-Luer-Lock注射器,6-过滤器,61-0.22um过滤器,62-0.45um过滤器。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
请参阅图1-3所示,培养装置由盒体(1)、阀门(2)、连接管(3)、Q-syte分隔膜密闭式无针接头(4)、Luer-Lock注射器(5)、过滤器(6)和密闭栓(7)组成;盒体(1)由上盒片和下盒片紧密贴合组成,上盒片和下盒片固定于刚性框架下,上盒片和下盒片中心部位向盒体(1)外侧凹入形成矩形容积腔(12),在盒体(1)的左右两侧各有一个变截面圆柱状通道,通道内固定有变截面圆柱状的母槽连接管(13);Luer-Lock注射器(5)的接头为螺旋接头。
试验步骤
1、材料
1.1试剂
1.2 装置
1.2.1 生物安全柜
1.2.2 37℃和5%CO2的孵化器
1.2.3 水浴
1.2.4 倒置显微镜
1.2.5 -80℃冷冻器
1.3 细胞
1.3.1 293T细胞
1.3.2 原代肝细胞
1.3.3 Huh7.5.1细胞
2、方法建立及注意事项
2.1一般方法
CCS系统由25cm2的盒片和连接装置组成,培养装置是一个紧密的细胞培养盒体,盒体由两个培养面组成,培养面固定在一个刚性框架上通过标准Luer-Lock连接口连接形成一个无菌的细胞培养腔体,培养表面是平的、透明的并具有透气性能。CCS系统用来培养293T细胞(LVs制品)和PPH(用于转导实验),该系统与使用OCS系统在10cm2的Petri盘体上培养细胞类似。在OCS系统中慢性病毒(LVs)和PPH培养上层清液中存在的VSV-G-pseudotyped粒子可以使用Huh7.5.1细胞检测出来。只有在CCS系统中培养实施步骤才可以细化。
2.2在CCS中管理液体转移
由于培养系统是封闭的,通过使用注射器推拉相同体积的液体或气体把所有的液体(细胞悬浮、培养介质、LVs等)从一个腔体(注射器,培养装置)传输到另外一个腔体中。此外,在操作之前还需要检查所有的阀门是打开的,避免培养装置内部的气压过高,检查连接末端的插头为0.22um的过滤器或者Q-syte注射部位。
为了注入液体,使用20mL的注射器吸入2mL气体然后吸入10mL液体制成一个空气存储注射器,这可以避免液体(尤其是细胞悬浮液) 留在试管中。通过倾斜培养装置另一面注射和推吸试管中的泡沫可以将泡沫挤出培养装置。注射超过10mL的液体将会导致多余的液体残留在试管中。
3.试剂制备
3.1转导质粒的制备
首先使用PCR对hTERT+p53DD、cyclinD1+CDK4R24C和c-mycT58A+HRasG12V进行增强处理,在CMVV5-LUC受体质粒上的XmaI和KpnI部位之间克隆三个插入序列,并对插入序列进行荧光素酶标记基因操作,得到荧光素标记的pLenti-hTERT+53DD、pLenti-cyclinD1+CDK4R24C和pLenti-c-mycT58A+HRasG12V质粒,使用荧光素酶pLenti-CMVV5作为控制荧光素酶报告基因的控制质粒;
3.2 293T细胞和Huh7.5.1细胞的细胞培养基(CM)制备
向细胞培养基中添加体积比为10%的灭活胎牛血清,体积比为1%的非必需氨基酸,10ug/mL的庆大霉素;使用0.22um的过滤器(61)过滤后在4℃的环境中保存最多两个月;
3.3原代肝细胞(PPH)培养基(WM)制备
向500mL的William’s E培养基添加10mL的质量体积比为7.5%的牛血清蛋白,5mL的ITS-G,10-7M的地塞米松溶液,体积比为1%的非必需氨基酸,体积比为1%的青霉素和链霉素的混合溶液,体积比为1%的稳定性谷氨酸;使用0.22um的过滤器过滤后保存在-80℃的环境中。
3.4原代肝细胞(PPH)离析
将肝切成小块组织立即置于含冰块的生理盐水中,在切割表面上插入可视装置,然后在pH为7.4和37℃的环境中使用500mL的缓冲液对肝组织进行灌洗,缓冲液中包含8.3g/L的NaCl、0.5g/L的KCl、2.4g/L的HEPES,0.19g/L的EGTA;使用包含100g的IV型胶原酶、3.9g/L的NaCl、0.5g/L的KCl、2.4g/L的HEPES和0.7g/L的CaCl2·H2O的400mL缓冲液在37℃的环境中进行第二次灌洗;使用IV型的胶原酶循环灌注,然后移除大块的肝组织,轻轻摇动存储在冰水中的包含有2.4g/L的HEPESD的HBSS缓冲液即可得到靶细胞;将含有靶细胞的悬浮液通过尼龙网过滤后在4℃的环境中冲洗三次;使用Trypan试剂排斥实验验证得到的肝细胞的可用性;立即使用合格的细胞或者冻结在-80℃的添加有10%的胎牛血清溶液中以代用;
4.在封闭的培养系统中制作慢性病毒上清的步骤
第0天
4.1培养装置的准备工作
4.1.1培养装置上阀门(2)的位置连接两端为公槽的连接管(3)到培养装置的阀门(2)上,连接0.22um的过滤器(61)的母槽端到连接管(3)上,打开所有的阀门(2);
4.1.2移除25cm2的培养装置的盖帽,给其母槽端连接一个阀门(2),给阀门(2)连接一个两端为公槽的连接管(3),给连接管连接一个Q-syte注射器(5)。
4.2 293T细胞的准备
4.2.1在一个50mL的试管中,重新悬浮在12mL的培养基中的3.0到7.5×106的293T细胞;
4.2.2给Luer-Lock注射器(5)适配一个针头,吸入2mL空气然后吸入10mL的细胞悬浮液;
4.3培养装置中第一次注射293T细胞;
4.3.1使用Betadin在高压无菌中净化Q-syte注射部位(4);
4.3.2给Q-syte注射部位(4)连接上Luer-Lock注射器(5);
4.3.3打开右边的所有阀门(2);
4.3.4稍微倾斜培养装置的左边,然后注射10mL细胞悬浮液;
4.3.5当细胞悬浮液接近左边的阀门(2)时,关闭阀门然后使用贮存有空气的Luer-Lock注射器(5)精确对应培养装置中细胞液的体积,这样细胞悬浮液可以全部进入培养装置(1)中;
4.3.6检查充满于培养装置(1)中的细胞悬浮液是否在进入或在左边和右边的连接管(3)中有残留;
4.3.7关闭阀门(2);
4.3.8断开并丢弃Luer-Lock注射器(5);
4.3.9使用Betadin净化Q-syte注射部位(4);
4.3.10将培养装置(1)平放在孵化器中,在37℃和CO2浓度5%的环境中孵化6-8小时;
4.3.11识别平躺面;
4.4在培养装置中第二次注射293T细胞;
4.4.1使用Betadin净化Q-syte注射部位(4);
4.4.2连接一个空的Luer-Lock注射器(5);
4.4.3打开左右阀门(2);
4.4.4吸入上层清液以移除可能没有吸附的过多的细胞;
4.4.5断开Luer-Lock注射器(5);
4.4.6重复4.2和4.3;
4.4.7在37℃和CO2浓度5%的环境中孵化24小时,返回到培养装置中,将上下面互调。
第一天
4.5冲洗293T细胞;
4.5.1使用Betadin净化Q-syte注射部位(4);
4.5.2连接一个空的Luer-Lock注射器(5);
4.5.3打开左右阀门(2);
4.5.4吸入上层清液以移除细胞;
4.5.5断开并丢弃Luer-Lock注射器(5);
4.5.6使用新的Luer-Lock注射器(5),注射并移除10mL培养基以冲洗培养装置(1),然后丢弃Luer-Lock注射器(5);
4.5.7使用新的注射器(5)注入10mL培养基;
4.5.8关闭阀门(2);
4.5.9断开和丢弃注射器(5);
4.5.10使用Betadin净化Q-syte注射部位(4);
4.5.11在37℃和CO2浓度5%的环境中孵化1小时。
4.6转染混合物
4.6.1在1.5mL的Eppendorf试管中添加500uL的无菌水、8.1ug的HIV-gap-pol质粒,8.1ug的pbabe-hTERT+p53DD,pbabe-cyclinD1+CDK4R24C或者pbabe-cmycT58A+HRasG12V质粒、2.7ug pMD2VSV-G-Env质粒、62uL的CaCl2 2M;
4.6.2在一个5mL的圆底聚丙烯试管中添加500uL的HBS 2x,在轻微旋转后使试管中的物质沉入管体,然后在室温条件下孵化20分钟,旋转后添加9mL培养基。
4.7转染293T细胞
4.7.1使用Betadin净化Q-syte注射部位(4);
4.7.2连接一个空的Luer-Lock注射器(5);
4.7.3打开左右阀门(2);
4.7.4吸入上层清液;
4.7.5断开并丢弃Luer-Lock注射器(5);
4.7.6将含有2mL气体和10mL的转染混合物(培养基中包含转染试剂盒质粒)的Luer-Lock注射器(5)连接到Q-syte注射部位(4);
4.7.7注射培养基;
4.7.8关闭阀门(2);
4.7.9断开并丢弃Luer-Lock注射器(5);
4.7.10使用Betadin净化Q-syte注射部位(4);
4.7.11在37℃和CO2浓度5%的环境中孵化一夜;
第二天
4.8第一批慢性病毒载体制品
4.8.1使用Betadin净化Q-syte注射部位(4);
4.8.2连接一个新的注射器(5);
4.8.3打开左右阀门(2);
4.8.4吸入上清液;
4.8.5断开并丢弃Luer-Lock注射器(5);
4.8.6使用心得Luer-Lock注射器(5),注射并移除10mL培养基以冲洗培养装置(1),然后丢弃Luer-Lock注射器(5);
4.8.7使用新的注射器注入10mL培养基;
4.8.8关闭阀门(2);
4.8.9断开并丢弃Luer-Lock注射器(5);
4.8.10使用Betadin净化Q-syte注射部位(4);
4.8.11在37℃和CO2浓度5%的环境中孵化一夜。
第三天
4.9收获第一批慢性病毒载体
4.9.1使用Betadin净化Q-syte注射部位(4);
4.9.2连接充满空气的注射器(5);
4.9.3在左边移除0.22um过滤器(61);
4.9.4连接0.45um的过滤器(62)的母槽端到连接管(3)的公槽端;
4.9.5连接过滤器的公槽端到公母槽连接管(3)的母槽端;
4.9.6连接第二个连接管的的公槽端到25cm2的培养装置(1)(称为培养二号)的母槽端;
4.9.7在二号培养装置(1),连接一个公槽连接管到阀门上;
4.9.8连接0.22um的过滤器(61)到连接管上;
4.9.9打开所有阀门(2);
4.9.10将一号培养装置上面反转到下面;
4.9.11使用注射器将空气推入一号培养装置中,以移除慢性病毒上清液从一号培养装置到二号中;
4.10慢性病毒的低温贮藏;
4.10.1关闭所有阀门(2);
4.10.2将二号培养装置上的所有连接管丢弃(3);
4.10.3关闭二号培养装置上的插管;
4.10.4在-80℃下将二号培养装置直接冻结;
4.11慢性病毒第二批产品;
4.11.1更换0.45um过滤器(62)并连接到一号培养装置;
4.11.2使用Betadin净化一号培养装置的Q-syte注射部位(4);
4.11.3将含有2mL气体和10mL的培养基的注射器连接到Q-syte注射部位;
4.11.4打开左右阀门(2);
4.11.5注入培养基;
4.11.6关闭阀门,插入0.45um的过滤器(62);
4.11.7丢弃注射器(5);
4.11.8在37℃和CO2浓度5%的环境中孵化24小时。
第四天
4.12收获第二批慢性病毒载体
4.12.1重复4.9和4.10步骤激活三号培养装置中的慢性病毒;
4.12.2丢弃一号培养装置;
5.一个密闭培养系统中原代肝细胞的慢性病毒转导步骤
第0天
5.1在培养装置中注入PPH
5.1.1在25cm2的培养装置里注入10×106PPH重新悬浮;
5.1.2将培养装置平放在37℃和CO2浓度5%的环境中孵化4小时;
5.1.3将培养装置反转过来使过剩的细胞吸附在第二面上;
5.1.4将培养装置平放在37℃和CO2浓度5%的环境中孵化4小时
5.2PPH转导慢性病毒;
5.2.1将包含收获慢性病毒载体(二号和三号培养装置,见4.9到4.12)的培养装置放置在37℃的水中解冻;
5.2.2使用注射器将上清液从四号培养装置中移除,步骤如4.5.1-4.5.4
5.2.3断开并丢弃注射器(5);
5.2.4丢弃四号培养装置0.22um过滤器(61);
5.2.5将四号培养装置的公槽连接管连接到二号或三号培养装置上;
5.2.6连接一个阀门到二号或者三号培养装置上的母槽端;
5.2.7连接一个公母槽连接管到阀门上;
5.2.8连接一个Q-syte注射部位(4)到连接管(3)上;
5.2.9使用Betadin净化二号或者三号和四号装置的两个Q-syte注射部位(4);
5.2.10连接充满空气的注射器到二号或三号培养装置的Q-syte注射部位(4);
5.2.11连接一个空的注射器到四号培养装置的Q-syte的注射部位(4);
5.2.12打开所有阀门(2);
5.2.13将空气从注射器(连接到二号或三号培养装置)推出,同时使用空的注射器吸入空气。以转移慢性病毒从二号或三号培养装置到四号培养装置中;
5.2.14关闭所有阀门(2);
5.2.15断开并丢弃注射器(5)和二号培养装置;
5.2.16四号培养装置插上插管;
5.2.17平放四号培养装置在孵化器中,孵化器置于37℃和CO2浓度5%的环境中孵化24小时。
第一天
5.3冲洗PPH
5.3.1将四号培养装置中的慢性病毒上清液丢弃到五号培养装置中,见步骤4.9(四号培养装置代替一号,五号培养装置代替二号)
5.3.2关闭阀门(2);
5.3.3丢弃五号培养装置和0.45um的过滤器(62)并将其更换为0.22um的过滤器(61);
5.3.4连接保护10mL的WM培养基注射到四号培养装置上;
5.3.5打开阀门(2);
5.3.6注入10mL的WM培养基;
5.3.7关闭阀门(2);
5.3.8断开并丢弃注射器(5);
5.3.9丢弃0.22um过滤器(61)并更换为0.45um过滤器(62);
5.3.10丢弃六号培养装置中的WM,如步骤4.9;
5.3.11丢弃培养和0.45um的过滤器(62)并更换为0.22um的过滤器(61);
5.3.12连接包含10mL的WM的注射器;
5.3.14注入10mL的WM;
5.3.15关闭阀门(2);
5.3.16断开并丢弃注射器(5);
5.3.17平放四号培养装置在孵化器中,孵化器设置37℃和CO2浓度5%的环境中48小时;
第三天
5.4 PPH激活
5.4.1将四号培养装置中的培养上清液转移到培养七号培养装置中,如步骤4.9(四号培养装置代替培养一号、七号培养装置代替二号);
5.4.2连接包含10mL的trpsin-EDTA的注射器;
5.4.3注入trypsin然后在倒置显微镜下监视细胞分离。保持注射器连接在培养装置上;
5.4.4吸入注射器中的细胞;
5.4.5断开注射器(5);
5.4.6转移50mL试管中的细胞并冲洗细胞以便以后使用;
5.4.7重复5.4.2到5.4.6以激活在培养中的细胞。
5.5在CCS中低温贮藏PPH
5.5.1将四号培养装置中的培养上清液转移到七号培养装置中,如步骤4.9(四号培养装置代替培养一号、七号培养装置代替二号);
5.5.2连接包含10mL的低温贮藏介质的注射器;
5.5.3注入低温贮藏介质(低温介质中添加有10%的FCS);
5.5.4断开注射器(5);
5.5.5在-80℃低温下冻结四号培养装置;
5.5.6使用前将细胞置于37℃的水中解冻;
5.5.7激活细胞如步骤5.4。
6动物试验
动物试验要获得当地伦理委员会(Comite′Re′gional d’Ethique en Matie`re d’Expe′rimentation Animale de Strasbourg;approval No.AL/01/18/08/12)的批准,然后在生物安全等级为3级的实验室中进行。
6.1注入转导的PPH
在50uL的WM中数量为1×106的转导PPH悬浮液通过皮下注射到NMRI-Foxn1nu/Foxn1nu小鼠的背部,注射前使用吸入异氟烷((Isoflo;Axience,Pantin,France)对小鼠全身麻醉
6.2生物荧光显影
转导PPH的荧光素酶活性在移植后指定的时间测定,测定时使用生物荧光显影技术,该技术在给小鼠腹腔内注射100uL的d-荧光素(20mg/mL;Caliper Lifesciences)后使用IVIS50的相机(caliper Lifesciences,Roissy,France)拍照。在20分钟内重复如前所述的1分钟内获得物,从荧光素注射的时间开始到整个荧光发射时间直到观察到信号开始降低。肿瘤细胞生物发光,可以使用动态显影软件3.1分析,可以表达为光子每秒每平方厘米每球面度(p/s/cm2/sr)在荧光素注射后导致生物发光到达最大值后使用时间点。通过计算标明的天数到移植后期的生物荧光比值来得到相关肿瘤细胞的生长。
7.质量控制
7.1转导效率
为了评估LVs的转导效率,50000个转导PPH在5.4.6步骤中激活,每个LV转导体被电镀在96式孔板上,然后在1250转速上离心5分钟。上层清液被移除,增加50uL的Glo Lysis(Promega,Madison,WI)。三分钟后加入25uL的BrightGlo荧光素(Promega)然后将25uL的混合物转移到一个不透明板测试荧光素酶的表达活性,检测时使用Mithras LB940 luminometer(Berthold,Thoiry,France)。相同数量的标准化PPH作为阴性对照组进行相似评价。
7.2 Huh7.5.2细胞的转导
为了评价LV转导后在PPH培养的上清液中LV产品的功效或者LVs重组体的复制能力,1×104的Huh7.5.1细胞被置于平底的96式碟子中,然后培养一夜。在PPH转导后,将CM以吸入的方式移除然后使用50uL的LV(覆盖来自步骤5.4.1的培养7号)或者三天内收获的培养上清液一式三份代替(覆盖来自步骤4.9或者4.2得到的培养2号或者3号)。在37℃和CO2浓度5%的条件下孵化6个小时后,添加100uL/well的CM,然后继续培养3天。然后移除上清液,添加50uL的lysis荧光素酶。相同数量的标准化PPH作为阴性对照组进行相似评价。
检测修复
8.封闭培养系统中得到LV产量
首先我们要评价CCS系统中荧光素酶编码控制LVs或者致癌基因编码的效率是否与常规的OCS系统更高。培养并使用质粒转染CCS系统中的293T细胞,或者在OCS系统中的PtriP培养皿。培养的上清液作为用以转导Huh7.5.1细胞,在OCS系统中操作进行以确保转导效率。转导Huh7.5.1细胞的荧光酶活性可以使用三天后的生物荧光活性进行量化。可以观察到从一个LV到另外一个的生物荧光活性水平有差别,这也反映了质粒结构对于LVs产品转导有不同的效率,对比CCS与OCS系统,转导效率对于每个独立的带菌体是相似的。结论表明使用CCS系统并不妨碍293T细胞转染产生LVs,该结论可从图4中分析得出。
9.在密闭系统中转导原代细胞
然后我们评价在CCS中转染原代细胞的可能性。对比CCS与OCS,使用控制LV或者每个致癌基因编码的LV转染独立地转染PPH。24小时后的初次转导,冲洗PPH然后在荧光素酶活性量化前再培养48小时。考虑到在CCS和OCS中转染,荧光素酶活性类似,这表明两个培养系统相同的转导效率,该结论可由图5分析得到。
在CCS中转导的PPH得到的组合体LV粒子制品复制能力的缺乏通过在第三天收获的PPH转导体中的Huh7.5.1细胞来评价。荧光素酶的活性在三天后量化后得到的结果与在没有转导PPH上清液中培养的Huh7.5.1细胞得到额结果类似,表明在CCS中通过PPH转导可以得到没有复制能力的组合体LV粒子,该结论可由图6分析得到。
10.在密闭培养系统中低温贮藏转导细胞
然后我们评价直接在CCS系统中低温贮藏转导细胞的可能性。在两个CCS系统中使用控制LV得到两个转导PPH个体。三天后,上清液移除并被低温贮藏基质取代。使用前将CCS装置直接冷冻并置于-80℃中一个月。为了证明它们的存活性,解冻的细胞立即通过皮下注射到免疫缺陷的NMRI nude小鼠体内,分别在5小时、48小时和6天检测生物荧光素活性。如图7所示,解冻的细胞荧光素酶活性可以在转移后第6天侦测到,该结果表明直接在CCS系统中冻结细胞的可行性。
在本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料过着特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为所述的具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
- 还没有人留言评论。精彩留言会获得点赞!