一种基因点突变修复的方法与流程
本发明涉及分子生物学基因编辑领域。具体而言,本发明用于通过基因编辑方法在哺乳动物细胞中对基因突变进行定点修复与外源基因敲入。
背景技术:
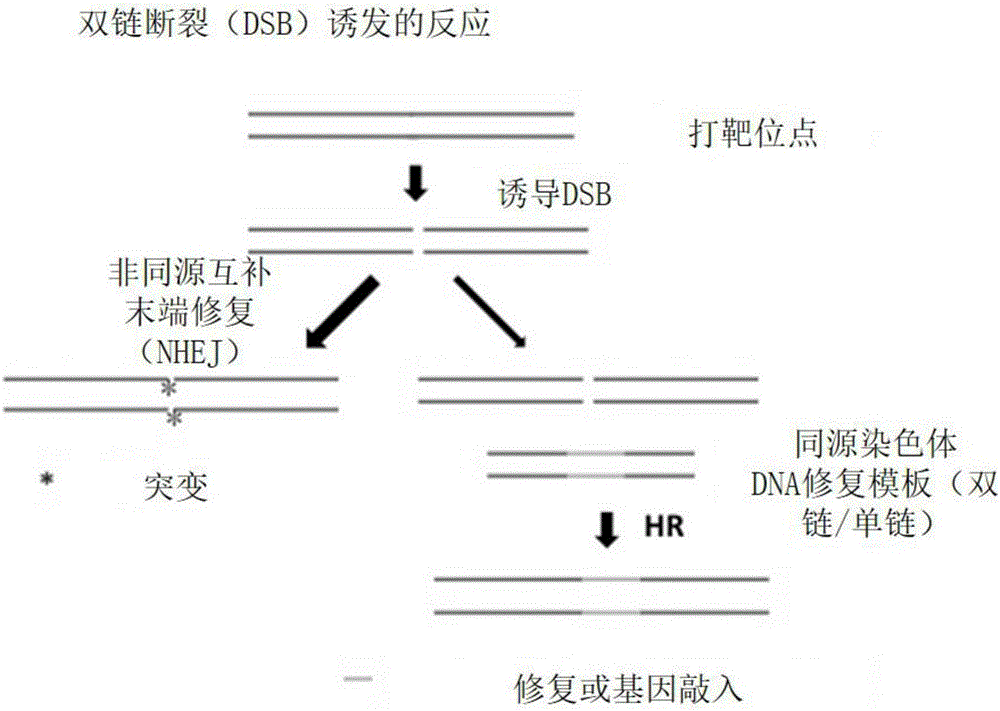
目前基因组定点编辑/修饰的基本原理是利用在靶位点区自发或诱发的DNA双链缺口(double-strain breaks,DSBs),DSBs将激活细胞内的DNA修复机制来进行基因组的改造,比如非同源区的末端连接(Non-homologous end joint,NHEJ)或是同源重组(Homologousrecombination,HR)(附图1)。
在哺乳动物细胞内,DSB自发产生的概率约低于1/104,如果通过基因工程方法采用spCas9和SaCas9等核酸酶诱发DSBs,效率可提高至10%以上,且具有位点特异性,因此方便了下一步对内源基因靶位点进行的基因修复过程顺利进行。DSBs激活细胞内的DNA修复通路后,会有两种不同的修复机制竞争性的参与DSBs的修复,一种是非同源区的末端连接NHEJ,一种是同源重组HR。要实现基因组靶位点的精准编辑,需要依赖胞内的同源重组修复机制。因此直接提高DSBs修复过程中同源重组发生的频率或抑制非同源区的末端连接(NHEJ)都有助于提高基因组定点编辑/修饰的效率。
非同源区的末端连接(NHEJ)和同源重组(HR)的发生都有多种蛋白参与其中,其中LIG4蛋白是NHEJ的重要因子,Rad51、Rad52和Brca1则是同源重组修复过程的重要参与蛋白。因此如果能够抑制LIG4蛋白的表达和/或促进Rad51、Rad52和Brca1的表达将有助于提高基因组定点编辑/修饰的效率。
Cas9等核酸酶对基因组的切割需要sgRNA的引导,当sgRNA的长度为20-21bp时,Cas9可以识别并切割靶位点,从而在靶位点处留下DSBs缺口;当sgRNA长度为14-15bp时,Cas9可以识别并结合靶位点,但却无法切割该位点。这一特性可被加以改造用于基因转录的抑制/激活。通过设计不同长度的sgRNA,我们还可以同时实现靶基因的激活/抑制和靶位点的切割与同源修复。
技术实现要素:
本发明提供了一种基于同源重组的基因编辑方法,包括采用核酸酶对目标基因进行切割,其特征在于:所述方法中还包括ssgRNA和靶向性转录调控因子,所述ssgRNA能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述靶向性转录调控因子能够调控基因编辑相关蛋白的转录。
其中,所述的核酸酶对目标基因进行同源重组包括采用sgRNA,所述sgRNA能够引导核酸酶靶向目标基因进行切割;优选所述sgRNA包括3'端的保守间隔相邻基序,优选所述的基序为NGG;优选所述sgRNA为18-23bp,优选20bp。
在本发明的一些具体实施例中,所述的核酸酶选自Cas9和CPF1,所述Cas9优选选自SaCas9、SpCas9。
在本发明的一些具体实施例中,所述方法中还包括同源互补修复模板,优选在同源重组的整合位点删除终止密码子。
在本发明的一些具体实施例中,所述的ssgRNA包括3'端的保守间隔相邻基序,优选所述的基序为NGG;优选所述的ssgRNA为14-16bp,更优选为14bp、15bp或16bp。
本发明利用DNA序列特异性的染色体双链DSBs生成技术,通过改造sgRNA并设计不同长度的sgRNA,辅以靶向性转录调控因子来实现高效、精确的基因组定点改造。在本发明的一些具体实施例中,所述的靶向性转录调控因子选自靶向性转录激活因子或靶向性转录抑制因子的至少一种,优选所述靶向性转录激活因子选自VP64,优选所述靶向性转录抑制因子选自KRAB。
本发明利用Cas9与不同长度sgRNA结合时活性的不同,实现对不同靶位点的差异调控,通过激活同源重组酶的表达/抑制NHEJ修复关键酶的表达,提高同源重组效率从而实现内源性基因突变的高效精准修复。在本发明的一些具体实施例中,所述的基因编辑相关蛋白选自非同源区末端连接相关蛋白和同源重组相关蛋白的至少一种,优选所述非同源区末端连接相关蛋白选自LIG4,优选所述同源重组相关蛋白选自Rad51、Rad52和Brca1或其组合。
本发明利用序列特异性的核酸酶(sequence-specific endonuclease)在靶位点处或附近制造一个DSBs;在此基础上,本发明利用长度不同的sgRNA和靶向性转录激活/抑制因子,激活同源重组相关蛋白表达,抑制非同源性的末端连接相关蛋白表达,实现靶位点处高效特异性的同源重组修复(见附图2,原理图)。
在本发明的一些具体实施例中,所述方法中的核酸酶与靶向性转录调控因子形成融合蛋白,优选所述融合蛋白包含柔性连接子。
在本发明的一些具体实施例中,所述的ssgRNA还偶联了RNA适配体序列,所述RNA适配体序列能够特异性的与关联蛋白结合,所述靶向性转录调控因子与所述的关联蛋白形成融合蛋白。
在本发明的一些具体实施例中,所述方法中的核酸酶与第一靶向性转录调控因子形成融合蛋白,优选所述融合蛋白包含柔性连接子,所述的ssgRNA还偶联了RNA适配体序列,所述RNA适配体序列能够特异性的与关联蛋白结合,第二靶向性转录调控因子与所述的关联蛋白形成融合蛋白。
本发明中,引入转录调控因子的策略可以但不限于,策略1:核酸酶与靶向性转录调控因子形成融合蛋白;或策略2:靶向性转录调控因子与所述关联蛋白(例如MS2结合蛋白或PCP结合蛋白)形成融合蛋白,再与偶联了RNA适配体序列(例如MS2或PP7)的ssgRNA结合;或策略3:在一个方法中同时存在上述两种策略。
在同源重组的基因编辑方法中,引入转录调控因子的策略还包括本领域技术人员根据现有技术或常规技术将转录调控因子(例如VP64或KRAB)引入所述同源重组相关蛋白(例如选自Rad51、Rad52和Brca1或其组合)启动子区的其他可能策略,例如通过融合蛋白的表达,转录调控因子表达在核酸酶-ssgRNA复合体上,只要所述转录调控因子具有能够激活或抑制所述同源重组相关蛋白的转录的作用。
另外,申请人在实验中发现,策略1和3与策略2的结果相似,这些基因编辑方法均可以大幅度提高了哺乳细胞中同源重组发生的效率。其中,策略3相比策略1和2可以更高效激活或抑制靶基因的转录。根据此原理,本领域技术人员设计靶向所述同源重组相关蛋白(例如选自Rad51、Rad52和Brca1或其组合)启动子区的ssgRNA并将转录调控因子(例如)引入所述同源重组相关蛋白(例如选自Rad51、Rad52和Brca1或其组合)启动子区的任何可能策略也属于本发明的发明内容。
在本发明的一些具体实施例中,所述RNA适配体为发夹结构;优选所述发夹结构选自MS2或PP7,所述关联蛋白选自MS2结合蛋白或PCP结合蛋白。例如本发明利用改造的sgRNA(携带有MS2/PP7等特殊的发夹结构,可被MS2结合蛋白/PCP蛋白等特异性识别),辅以靶向性转录激活/抑制因子(MS2/PCP-VP64/KRAB),可以激活/抑制特定基因的转录。
另一方面,本发明还提供了一种用于基因编辑的组合物,包括ssgRNA和靶向性转录调控因子,所述ssgRNA能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述靶向性转录调控因子能够调控基因编辑相关蛋白的转录。
其中,所述组合物还包括核酸酶、sgRNA、同源互补修复模板或其组合。
在本发明的一些具体实施例中,本发明提供了一种用于基因编辑的组合物,包括核酸酶、核酸酶与靶向性转录调控因子形成的融合蛋白、sgRNA、ssgRNA和同源互补修复模板,所述ssgRNA能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述靶向性转录调控因子能够调控基因编辑相关蛋白的转录。
在本发明的一些具体实施例中,本发明提供了一种用于基因编辑的组合物,包括提供核酸酶、sgRNA、RNA适配体序列与ssgRNA的偶联分子、同源互补修复模板、靶向性转录调控因子与RNA适配体关联蛋白的融合蛋白,所述ssgRNA能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述靶向性转录调控因子能够调控基因编辑相关蛋白的转录。
在本发明的一些具体实施例中,本发明提供了一种用于基因编辑的组合物,包括提供核酸酶与第一靶向性转录调控因子形成的融合蛋白、sgRNA、RNA适配体序列与ssgRNA的偶联分子、同源互补修复模板和第二靶向性转录调控因子与RNA适配体关联蛋白的融合蛋白,所述ssgRNA能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述靶向性转录调控因子能够调控基因编辑相关蛋白的转录。
在本发明的一些具体实施例中,所述的靶向性转录调控因子选自靶向性转录激活因子或靶向性转录抑制因子的至少一种,优选所述靶向性转录激活因子选自VP64,优选所述靶向性转录抑制因子选自KRAB。
在本发明的一些具体实施例中,所述的基因编辑相关蛋白选自非同源区末端连接相关蛋白和同源重组相关蛋白的至少一种,优选所述非同源区末端连接相关蛋白选自LIG4,优选所述同源重组相关蛋白选自Rad51、Rad52和Brca1或其组合。
另一方面,本发明还提供了一种RNA适配体序列与ssgRNA的偶联分子,所述的ssgRNA部分能够引导核酸酶靶向基因编辑相关蛋白的编码核酸但不会发生切割,所述RNA适配体序列部分能够特异性的与关联蛋白结合;优选所述的ssgRNA部分包括3'端的保守间隔相邻基序,优选所述的基序为NGG;优选所述的ssgRNA部分为14-16bp,优选为14bp、15bp或16bp。
在本发明的一些具体实施例中,所述RNA适配体为发夹结构;优选所述发夹结构选自MS2或PP7,所述关联蛋白选自MS2结合蛋白或PCP结合蛋白。
另一方面,本发明还提供了一种试剂盒,包括本发明中所述的任一种所述的组合物或任一种所述的偶联分子。
另一方面,本发明还提供了本发明中任一种所述的基因编辑方法或任一种所述的组合物或任一种所述的偶联分子用于基因编辑的用途。
在本发明中,所述的基因来自于微生物、植物、动物、细胞、哺乳动物或人。
本发明的有益效果
本发明大幅度提高了哺乳细胞中同源重组发生的效率;
本发明在不导入外源基因的情况下提高了同源重组效率,能够避免现有的提高同源重组技术例如小分子化合物等易于产生染色体组不稳定性等缺点。
本发明提供了在哺乳动物中进行准确、精细基因编辑的方法。
本发明提供了可在哺乳动物里同时对多个基因进行突变修复的方法。
附图说明
图1示出的是DSB介导的基因修饰方法的图
图2示出的是体系示意的图。
a.当ssgRNA长度为14bp时,Cas9不具备核酸酶活性。通过三种策略可以实现14bp ssgRNA对特定基因的转录调控:策略一,构建Cas9-VP64/KRAB的融合蛋白,辅以14bp ssgRNA可以激活/抑制靶基因的转录;策略二,使用野生型Cas9,在ssgRNA序列中添加MS2/PCP等RNA适配体序列,再辅以MS2/PCP-VP64/KRAB融合蛋白,可以激活/抑制靶基因的转录;策略三,构建Cas9-VP64/KRAB的融合蛋白,在ssgRNA序列中添加MS2/PCP等RNA适配体序列,再辅以MS2/PCP-VP64/KRAB融合蛋白,可以更高效的激活/抑制靶基因的转录。根据此原理,设计靶向LIG4/RAD51/RAD52/BRCA1的ssgRNA,可以调控系列基因的表达。
b.在具体实施中,本发明的方法被应用到GFP报告基因的修复(详见实验流程),外源的GFP表达载体中GFP编码区存在终止密码子而无法翻译生成绿色荧光蛋白。只有精确的定点修复,才有可能实现预期的GFP表达,而通过流式细胞分选检测GFP阳性细胞的比例,可以定量定点修复的效率。本方法还被应用到U251人胶质瘤细胞系中內源TP53基因突变的修复,通过细胞转染、培养、提取细胞基因组和PCR测序的方式,可以对基因突变定点修复的效率进行检测(详见实验流程)。
图3示出的是敲入载体构建的图
图4示出的是基因编辑效率比较的图
图5示出的是BFP稳定转染细胞系中基因突变修复效率比较
图6示出的是內源TP53基因突变修复效率比较
具体实施方式
下面通过具体实施方式及实验数据对本发明作进一步的说明。尽管为了清楚的目的,在下文中使用了专用术语,但这些术语并不意味着定义或限制本发明的范围。
在本发明中,术语“靶位点”即在目标基因组中任何一段欲加以改造或修复的DNA序列。靶位点附近的DNA序列,允许外源序列在靶位点处的整合,包括但不限于基因敲入(knock-in)。在具体实施方式中,目标DNA序列是双链的DNA序列,包括,但不限于,细胞的染色体基因组中的DNA序列、细胞染色体基因组外的DNA序列(例如线粒体基因组)、质粒、病毒等的DNA序列。
在本发明中,术语“定点重组”是指,将外源序列通过非随机的方式整合到特定的靶位点处,包括整合到某特定靶位点的5’上游、3’下游或者靶位点之间。
在本发明中,术语“外源DNA序列”是指,期望被定点重组到靶位点处的DNA序列。外源DNA序列可以是靶位点处不存在或被改变的序列。
在本发明中,“sgRNA”是指向导RNA,可引导核酸酶靶向目标基因进行切割;所述sgRNA可以包括3'端的保守间隔相邻基序,例如基序为NGG;所述sgRNA的长度可以为18-23bp之间。
在本发明中,“ssgRNA”是指长度为14-16bp的向导RNA,在ssgRNA的引导下,Cas9可以识别并结合靶位点,但却无法切割靶位点。
在本发明中,“适配体”、“适体”、“结合适体(aptamer)”具有相同含义,是指是经过一种体外筛选技术(systematic evolution of ligands by exponential enrichment,SELEX),从随机单链寡聚核苷酸文库中得到的能特异结合蛋白或其他小分子物质的单链寡聚核苷酸,也可以是RNA,也可以是DNA,长度一般为25~60个核苷酸。
具体实施例
实施例1GFP报告基因改造,恢复GFP绿色荧光蛋白的表达
1)改造ssgRNA,获得带有特定RNA发夹结构的ssgRNA
在ssgRNA(SpCas9和SaCas9)的特定位点接入独特的RNA发夹如MS2/PP7,这类发夹结构可被MS2结合蛋白/PCP蛋白所特异性识别。
有PP7发夹结构的ssgRNA全序列为:
ccggtgagaccgagagagggtctcagttttagagctagcgagggagcagacgatatggcgtcgctccctcgttagcaagttaaaataaggctagtccgttatcaacttgcgagggagcagacgatatggcgtcgctccctcgtaagtggcaccgagtcggtgctttttgaattc
带有MS2发夹结构的ssgRNA全序列为:
ccggtgagaccgagagagggtctcagttttagagctaggccaacatgaggatcacccatgtctgcagggcctagcaagttaaaataaggctagtccgttatcaacttggccaacatgaggatcacccatgtctgcagggccaagtggcaccgagtcggtgcttttttt
以上ssgRNA全序列均在南京金斯瑞生物科技有限公司合成。
2)GFP基因靶位点处潜在的Cas9识别位点分析
本实验中,GFP报告基因改造的目标是替换终止密码子,恢复GFP绿色荧光蛋白的表达。对应的转基因的操作是在GFP编码区终止密码子附近引入DNA双链断裂(DSBs),之后通过同源修复模板加以修复。因此首先必须于靶位点区即GFP编码区终止密码子附近确定整合位点。在本发明方案中,即选定Cas9识别位点,整合位点可选在识别位点中间,以达到破坏识别位点,保证整合序列不被继续切割。
如上所言,在特定DNA序列上有效产生DSBs可以有多种策略,包括基因工程改造过的I-sceI、I-AniI、FoxI、Cas9以及一些合成多核苷酸,如LNA、PNA等。本实验采用spCas9和SaCas9,与配套的改造过的ssgRNAs联用。根据序列分析,发现GFP编码区终止密码子附近有潜在的spCas9识别位点(序列如下):
GFP终止密码子(ATG)附近序列图,标注识别位点
ATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCtagGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGC
小写字母tag为终止密码子
斜体字母区域为spCas9识别位点,划横线区域“agG”为PAM序列
3)设计同源互补修复模板(donor DNA template)
同源互补修复模板设计如图3,在ssgRNA靶位点的上下游各有100-200bp的同源序列(上下游同源臂)。在整合位点处删除终止密码子序列。整体连入T载体经过扩增提取质粒成为外源供体。
外源供体构建体:
100bp上游同源重组臂—100bp下游同源重组臂
具体序列为:
gacgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgtttttgagctcccaacgcg
4)构建MS2/PCP-VP64/KRAB融合蛋白表达载体
为将转录调控因子准确定位至DSBs修复相关的基因位点,须在ssgRNA中连入特定的发夹结构如MS2或PP7,之后构建MS2/PCP与转录调控因子如VP64或KRAB的融合蛋白表达载体,此处MS2蛋白可识别ssgRNA中的MS2发夹结构,PCP则可以识别PP7发夹结构。为此构建MS2/PCP-VP64/KRAB融合蛋白表达载体,将MS2/PCP的编码序列分别与VP64和KRAB融合,连接在带有EF1alpha启动子的表达载体中,保证读码框一致可读通。
5)转染293细胞系确定DNA模板的修复效率
实验组采用带有终止密码子突变的GFP表达载体,spCas9表达载体,GFP特异性的sgRNA(20bp),带有MS2发夹结构靶向LIG4/RAD52/RAD51/BRCA1的ssgRNA(14bp)、MS2-KRAB(对应LIG4)/VP64(对应RAD52/RAD51/BRCA1)融合蛋白通过Lipofectamine 3000共转293细胞系,37℃培养箱培养48小时后,通过流式细胞分选(flow cytometry)检测GFP阳性细胞占总细胞的比例。
对照组中同样采用相同量的带有终止密码子突变的GFP表达载体,spCas9表达载体,GFP特异性的sgRNA(20bp),带有MS2发夹结构无靶向的ssgRNA、MS2-KRAB(对应LIG4)/VP64(对应RAD52/RAD51/BRCA1)融合蛋白通过Lipofectamine 3000共转293细胞系,37℃培养箱培养48小时后,通过流式细胞分选(flow cytometry)检测GFP阳性细胞占总细胞的比例(图4)
我们利用点突变的BFP构建了稳定转染细胞系,通过BFP点突变的修复可产生GFP绿色荧光蛋白对修复的效率做定量的统计。我们发现抑制NHEJ通路Lig4可以显著提高同源重组修复的效率(图5左)。而促进同源重组修复的通路组分Rad51/52也可显著促进同源重组修复的效率(图5右)。提示如将抑制Lig4表达与促进Rad51/52表达联合使用则可以进一步提升基因突变同源重组修复效率。
实施例2人TP53基因点突变修复的实验设计
1)选用U-251人胶质瘤细胞系,测序结果显示该细胞系的TP53基因携带有R273H(CGT->CAT)纯合点突变。为修复该点突变,在该位点附近设计特异性靶向TP53基因的sgRNA(SaCas9)(图6)。
其具体序列:
agagaccggcgcacagaggaa
PAM:gagaat
2)设计修复模板:模板选用DNA单寡核苷酸链,序列为actgggacggaacagctttgaggtgcgtgtttgtgcctgtcctgggcgtgataggcgaacggaagaggaaaacctccgcaagaaaggggagcctcaccacgagctgcccccagggag。TP53的sgRNA识别位点处引入同义突变以避免修复后的位点被SaCas9继续切割而引入插入/缺失突变。
3)转染U251细胞系验证突变修复效率:实验组中转入携带有TP53特异性靶向sgRNA的SaCas9表达载体、同源修复模板、puromycin筛选质粒和修复因子表达载体(带有MS2发夹结构靶向LIG4/RAD52/RAD51/BRCA1的ssgRNA(14bp)、MS2-KRAB(对应LIG4)/VP64(对应RAD52/RAD51/BRCA1)融合蛋白),对照组中转入携带有TP53特异性靶向sgRNA的SaCas9表达载体、同源修复模板、puromycin筛选质粒和对照载体。通过PEI共转U-251细胞系,37℃培养箱培养48小时后,加入2ug/ml的puromycin继续培养48小时,提取细胞基因组,采用TP53特异性引物通过PCR实验扩增修复区域,送测序分析修复效果(图6,因子1:Rad51+MS2-VP64;因子2:Rad52+MS2-VP64;因子3:Brca1+MS2-VP64;因子4:LIG4+MS2-KRAB)。
4)实验结果分析
通过对外源GFP突变基因的修复系统测试,我们发现Rad52重组酶可有效的提高同源重组效率至50%以上(参见图4),此结果将大大增加基因突变的修复效率,此研究内容及思路尚无任何国际研究论文发表,属国际首创的发现。
在第二部分对内源癌基因的突变修复实验中,我们进一步验证,Rad52可有效促进TP53基因突变的修复,此修复测试实验尚未用流式细胞分选阳性细胞,即用PCR测序的方法得到了非常均一的突变修复测序峰图,说明修复效率很高,可达到体内修复突变癌基因以及其他在体基因突变修复及基因编辑的要求。
以上,基于本发明的实施方式进行了说明,但本发明不限定于此,本领域的技术人员应该明白,在本发明的主旨的范围内能够以进行变形和变更的方式实施,这样的变形和变更的方式,理应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!